Article Text

Statistics from Altmetric.com
In recent years it has been established that inflammation has a pathogenic role in atherosclerosis. Experimental studies have suggested that altering transcription of proinflammatory genes can result in the inhibition of atherosclerotic disease progression. Peroxisome proliferator activated receptor γ (PPARγ), a member of the nuclear receptor superfamily of ligand activated transcription factors, is highly expressed in several organs as well as in atherosclerotic plaques. Agonists of this receptor, such as rosiglitazone, pioglitazone, and troglitazone, have insulin sensitising actions and the former two agents are used clinically to treat type II diabetes. PPARγ agonists can also inhibit the transcription of proinflammatory genes within plaques and have antithrombotic effects. Furthermore, PPARγ agonists have been shown to inhibit vascular smooth muscle cell (VSMC) proliferation, which underlies restenosis after percutaneous coronary intervention. This article summarises our current understanding of the role of PPARγ agonists in atherogenesis and discusses their potential role in the treatment of coronary artery disease and its complications.
Inflammation and coronary atherosclerosis
It is well established that an inflammatory process and specific immunological mechanisms underlie atherosclerotic plaque formation and fibrous cap disruption.1 It may therefore be speculated that antagonising key proinflammatory processes may result in delay or inhibition of disease progression.
One of the earliest physiological changes in atherosclerotic disease is reduced production of nitric oxide by the vascular endothelium (that is, endothelial dysfunction) in response to pharmacological or haemodynamic stimuli.2 Nitric oxide has several protective roles including reduction of vascular tone, inhibition of platelet aggregation, and reduction of VSMC proliferation. The earliest pathological change in atherosclerosis is the accumulation of plasma low density lipoprotein in the subendothelial space and its oxidation.3 It is still unclear whether endothelial dysfunction is a cause or a consequence of subendothelial lipid accumulation. Oxidised low density lipoprotein activates endothelial cells, which in turn synthesise surface bound molecules (that is, selectins and adhesion molecules) and chemokines (for example, monocyte chemoattractant protein 1) that attract circulating monocytes and T cells and facilitate their migration into the subendothelial space.4 Here, the activated monocytes mature into macrophages and express the necessary scavenger receptors to ingest modified lipids and become foam cells.5 VSMCs also have an important role in the atherosclerotic plaque. Normally they reside in the vascular media where they act to maintain vascular tone. In atherosclerosis VSMCs migrate out to the site of injury where they have a reparative role. VSMCs produce growth factors that facilitate their proliferation at the site of injury. In addition, they produce certain matrix proteins (collagens and elastins) to repair the vessel and produce a fibrous cap. The fibrous cap separates the thrombogenic lipid rich core from circulating platelets and procoagulation factors and gives structural stability to the atherosclerotic plaque. Acting through diverse mechanisms, inflammatory cells can weaken the fibrous cap and make them prone to rupture. T lymphocytes, present in atheromatous plaques, synthesise interferon gamma, which inhibits VSMC proliferation and collagen synthesis, thus contributing to weakening of the fibrous cap.6 Interferon gamma also activates macrophages, which in turn produce metalloproteinases (for example, matrix metalloproteinase 9) that degrade the matrix proteins in the cap.1 Activated macrophages also produce proinflammatory cytokines such as interleukin 1β, tumour necrosis factor α, and interferon gamma that are cytotoxic for VSMCs. Overall, the balance between a subendothelial inflammatory stimulus and the local healing response of the VSMC determines the stability of the plaque. Plaques with a preponderance of inflammatory cells, a thin fibrous cap, and a paucity of VSMC have a tendency to fissure or to show endothelial erosions, thus exposing the thrombogenic plaque constituents.7 This facilitates platelet adhesion and aggregation, which may result in clinical events such as unstable angina or myocardial infarction.7
Several regulatory pathways have been identified that control the expression of atherogenic gene products. These include the transcription factor nuclear factor κB, which is involved in the activation of many genes involved in immune and inflammatory responses. These genes code for proinflammatory cytokines, chemokines, immune receptors, and adhesion molecules.8 Nuclear factor κB functions together with other transcription factors, such as activator protein 1 and signal transducers, and activators of transcription.8 From a therapeutic perspective, agents that may modulate the actions of these transcription factors would reduce the proinflammatory stimulus and potentially inhibit or delay atherosclerotic disease progression.
Insulin resistance, atherosclerosis, and the role of PPARγ agonists
Insulin resistance and hyperinsulinaemia are major risk factors for coronary heart disease, namely obesity, type II diabetes, dyslipidaemia, hypertension, atherosclerosis, and a procoagulant state. These risk factors constitute the insulin resistance syndrome and contribute to the increased risk of atherosclerotic disease seen in patients with insulin resistance or type II diabetes.9 The syndrome is also associated with endothelial dysfunction and likely causal mechanisms include dyslipoproteinaemia, increased oxidative stress, and advanced glycation end products.10
PPARγ heterodimerise with retinoid X receptor and alter the transcription of numerous target genes. Thiazolidinediones such as troglitazone, pioglitazone, and rosiglitazone are oral antidiabetic compounds that bind to PPARγ with high affinity. In adipocytes, PPARγ agonism by thiazolidinediones increases insulin responsiveness and their capacity for glucose uptake and lipid storage.11Thiazolidinediones increase glucose disposal in skeletal muscle and reduce hepatic glucose output.11 Genetic studies in humans show that PPARγ mutations can lead to severe insulin resistance.12
In the insulin resistance syndrome, thiazolidinediones not only improve glycaemic control but also have been shown to modify other components of the syndrome and potentially reduce cardiovascular risk. Experimental evidence shows that PPARγ agonists can reverse endothelial dysfunction and ameliorate both hypertension and the atherogenic lipid phenotype, two features of the insulin resistance syndrome.13-15 Patients with the insulin resistance syndrome and type II diabetes also have an abnormal fibrinolytic state evidenced by increased concentrations of fibrinogen and plasminogen activator inhibitor 1 (PAI-1). There is evidence that PPARγ agonists, by correcting insulin resistance, can reduce PAI-1 and fibrinogen concentrations and so improve fibrinolysis.16 Their potential benefit as inhibitors of atherogenesis in diabetic patients has been shown by a single study to date. This showed an inhibitory effect of three months of treatment with troglitazone on carotid intimal medial thickening.17 However, troglitazone has since been withdrawn from clinical use because of hepatotoxicity problems. Newer thiazolidinediones, such as rosiglitazone or pioglitazone, do not share the same risk of hepatotoxicity and are licensed for use in the treatment of type II diabetes.
There is reasonable evidence to suggest that, by ameliorating insulin resistance and its metabolic sequelae, thiazolidinediones can retard atherogenesis and disease progression. However, through direct inhibition of inflammatory processes within the vessel wall, thiazolidinediones may also inhibit atherosclerosis progression independently of any insulin sensitising action. Thus, its potential beneficial effects may not be restricted to type II diabetic patients but may have wider applications.
PPARγ agonists in inflammation, atherosclerosis, and thrombosis
PPARγ is expressed in endothelial cells, VSMCs, and macrophage cells within atherosclerotic plaques.18 Here it modifies transcription factors, such as nuclear factor κB, and inhibits activation of many proinflammatory genes responsible for plaque development and maturation.19 PPARγ agonists have been shown to inhibit production of proinflammatory cytokines such as tumour necrosis factor α, interleukin 1β, and interleukin 6.20 In addition they inhibit expression of the chemokine monocyte chemoattractant protein 1 and inflammatory enzymes such as inducible nitric oxide synthase.21 ,22 PPARγ agonists reduce the production of metalloproteinases by activated plaque macrophages, thus promoting plaque stability.23 Crucially, PPARγ agonists also reduce adhesion molecule expression in endothelial cells, thereby reducing macrophage homing to plaques in vivo.24 Thiazolidinediones can also inhibit endothelin 1 production in endothelial cells.25 Endothelin 1, a powerful vasoconstrictor peptide, regulates endothelial function and promotes smooth muscle cell proliferation. High concentrations of endothelin 1 correlate with increased severity of coronary artery disease.26 PPARγ activation also mediates inhibition of T cell responses.27 Thiazolidinediones may also have antithrombotic effects as they reduce PAI-1 production by endothelial cells in vitro.28
This general anti-inflammatory and antithrombotic role of PPARγ initially appeared to be in contradiction with its function in macrophage foam cells. It had been shown that monocytes when exposed to oxidised low density lipoprotein express PPARγ. Activation of PPARγ promotes monocyte differentiation and expression of the scavenger receptor CD36, which in turn facilitates increased uptake of oxidised low density lipoprotein.29 ,30 This cycle would promote foam cell formation and atherogenesis. However, there is recent evidence to show that in addition to lipid uptake, PPARγ promotes cholesterol efflux from the macrophage foam cell.31 This study showed that the net effect of this dual action is in fact lipid removal from the arterial wall.31
The multiple mechanisms by which PPARγ activation may potentially retard atherosclerotic disease progression are summarised in fig 1. Moreover, studies using animal models of atherosclerosis consistently show that PPARγ ligands reduce the extent of atherosclerosis.31-33

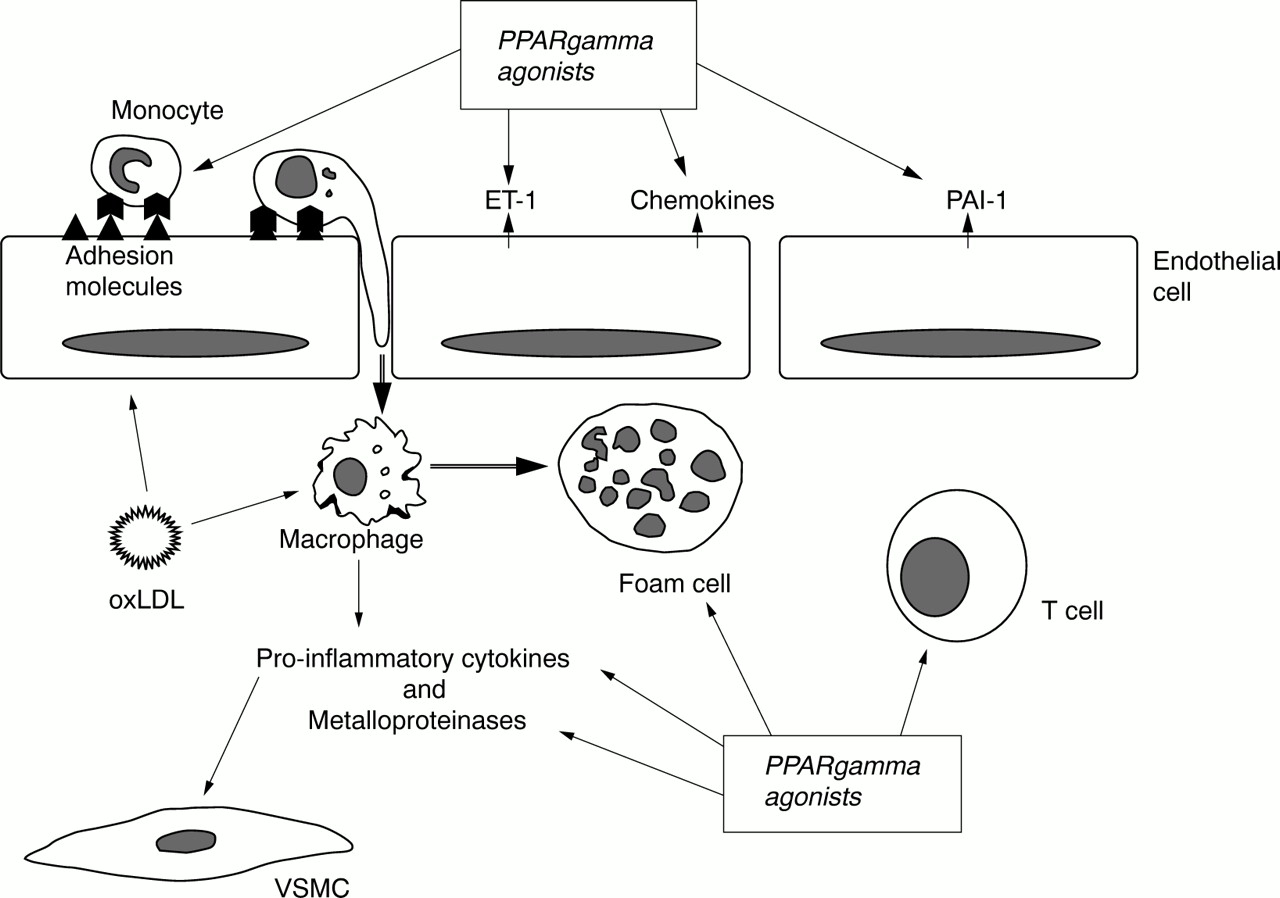{kind=link}
Schematic representation of sites of action of peroxisome proliferator activated receptor γ (PPARγ) agonists within atherosclerotic plaques. ET-1, endothelin-1; oxLDL, oxidised low density lipoprotein; PAI-1, plasminogen activator inhibitor 1; VSMC, vascular smooth muscle cell.
PPARγ agonists and restenosis after coronary angioplasty
Restenosis after successful percutaneous coronary intervention remains a major limitation of this technique and typically occurs 3–6 months after the intervention. Inflammation and response to injury are implicated in the pathogenesis of restenosis after percutaneous coronary intervention. Restenosis can occur as a result of a combination of three processes: firstly, elastic recoil of the vessel, which can occur within hours of the percutaneous coronary intervention; secondly, inadequate compensatory enlargement of the vessel after angioplasty (inadequate vascular remodelling), which may occur over several days; and thirdly, neointimal hyperplasia, secondary to VSMC proliferation and extracellular matrix synthesis.34 This phenomenon begins immediately after vascular injury and is induced by platelet and leucocyte adhesion.35 The release of vasoconstricting agents (for example, serotonin) and growth factors (for example, platelet derived growth factor) from platelets and injured VSMCs activates medial VSMCs. This stimulates VSMC proliferation and migration into the intima. The use of coronary stenting can reduce elastic recoil and inadequate remodelling, which accounts for their favourable impact on restenosis.36Stenting has reduced the incidence of restenosis from 40% to approximately 20% in published studies.37 However, stenting does not reduce neointimal hyperplasia mediated by VSMC proliferation, which is the primary mechanism of restenosis after stenting. Intrastent restenosis is a difficult problem in interventional cardiology and the best management remains unclear. The only effective treatment for the prevention of intimal hyperplasia is brachytherapy, and this technique raises concern over safety and practicality.38
Recent work, however, suggests that thiazolidinediones may reduce intimal hyperplasia after percutaneous coronary intervention and so prevent restenosis. Thiazolidinediones can inhibit VSMC proliferation by several mechanisms that may be beneficial in both diabetic and non-diabetic patients undergoing percutaneous coronary intervention. Diabetic patients in particular have a higher incidence of restenosis.39 Hyperglycaemia is known to increase VSMC migration and a study has shown that thiazolidinediones can inhibit this phenomenon.40 In addition, the abnormal fibrinolytic state in insulin resistant patients may promote restenosis. High concentrations of PAI-1 after percutaneous coronary intervention can predict who is at greatest risk of restenosis.41 As outlined previously, thiazolidinediones can reduce increased PAI-1 concentrations associated with insulin resistance.16
PPARγ ligands can also inhibit VSMC migration mediated by mechanisms other than insulin sensitisation. Matrix metalloproteinase 9 appears particularly important in promoting migration of VSMCs after arterial injury. PPARγ agonists were shown to reduce matrix metalloproteinase 9 expression and thereby inhibit VSMC migration.42 In addition, thiazolidinediones can inhibit angiotensin II and platelet derived growth factor induced VSMC proliferation in vitro.43 ,44 Animal studies, in diabetic and non-diabetic models, have found an inhibitory effect of thiazolidinediones on postinjury intimal hyperplasia.45 ,46 One study so far has examined the effects of troglitazone on restenosis in humans. This was an intravascular ultrasound study limited to type II diabetic patients who underwent coronary stenting. The study investigators reported a significant reduction in intimal hyperplasia in those treated with troglitazone.47
Summary
Recent work has suggested that PPARγ agonists are potential antiatherogenic agents. These compounds, because of their insulin sensitising action, are already used therapeutically in type II diabetes. However, they may also have direct antiatherogenic effects that may benefit all patients with coronary artery disease. To date there have been very few studies in patients and they have been confined to diabetic patients. Clearly further studies in both diabetic and non-diabetic patients are warranted to determine the true value of PPARγ agonists in the treatment of coronary artery disease.