Article Text

Statistics from Altmetric.com
Diastolic left ventricular disease is being increasingly incriminated as a cause of limitation of exercise tolerance, whether or not ejection fraction is normal,1,2 though the mechanisms by which it does so are far from clear. Indeed, it has been suggested that no diastolic abnormality at all need be demonstrated for a diagnosis of possible or probable diastolic heart failure to be made.3
WHAT IS DIASTOLIC FUNCTION?
Measurements can be made during diastole with many techniques, old or new, but can there be said to be as many impaired “functions” as there are abnormal measurements? Surely, the term “diastolic function” applies only to a small number of more basic mechanisms whose nature must be elucidated independently of the method used to detect them and whose number depends on rigorous use of Occam's razor. Difficulties in defining diastolic heart failure strongly suggest that agreement in this field has still to be achieved. Indeed, no simple definition of diastolic disease itself has emerged. “Increased resistance to filling” has been suggested. While the resistance of a valve orifice or circulation can readily be defined in terms of pressure drop and flow, resistance to filling involves neither and so is poorly defined. This lack of gold standards by which discrete mechanisms can be assessed in individual patients is a major impediment to identifying and quantifying disturbances in disease.
DEFINITION OF DIASTOLE
Left ventricular diastole is traditionally defined as the period in the cardiac cycle from the end of aortic ejection until the onset of ventricular tension development of the succeeding beat.4 Even in the normal subject, several mechanisms are involved:
Decline of the myocardial active state following systole.
Passive effects of connective tissue. Compression or extension of connective tissue may store potential energy from systole and release it in early diastole. In late diastole, the properties of connective tissue determine ventricular compliance.
Rapid changes in atrial and ventricular pressures.
Transmitral flow.
Interactions from right ventricle and pericardium.
Atrial systole.
Furthermore, even the traditional definition of diastole has been questioned, involving as it does arbitrary relations between muscle function and the circulation. Brutsaert5 has very properly suggested that from the point of view of muscle function, systole should be taken to include the entire period over which ventricular muscle develops tension, relegating diastole to the remainder of the cardiac cycle. Since the traditional definition is more familiar in the clinical context, it will be used here.
Diastolic function differs in important ways from that of systole:
It is much more age dependent, so that values that are normal in the young may be pathological in the elderly. “Diastolic heart failure” is a disease of the elderly.
Abnormal diastolic measurements do not necessarily reflect intrinsic diastolic disease, as they are greatly affected by abnormalities of ventricular activation and systole. Disturbances whose most obvious effects are seen in diastole may originate earlier in the cardiac cycle.
Diastolic measurements are critically dependent on ventricular filling pressure, which is likely to be abnormal in patients with pulmonary congestion.
Furthermore, the most effective means of treating heart disease do not affect ventricular myocardium directly, but modify left atrial and aortic pressures. Diastolic measurements may thus become more abnormal with treatment known to increase exercise tolerance or prognosis.
VENTRICULAR RELAXATION
The first problem in assessing relaxation is to define it. This is not self evident, and the word is used in several senses in the literature. We define it as the decline in myocardial tension caused by net uptake of calcium into the sarcoplasmic reticulum. It has no easily definable onset, because it is simply the terminal phase of the systolic active state.
Key determinants of diastolic function
-
Extrinsic
Ventricular activation
Age
Systolic function
Incoordination
Filling pressure
-
Intrinsic
Passive ventricular stiffness
Primary disturbances of relaxation are uncommon
Its time course is profoundly affected by events during systole, particularly the pressure load to which the myocardium has been subject.6
The most striking clinical manifestation of relaxation is the fall in left ventricular pressure. When recorded with a tip manometer, this fall is approximately exponential over the period during which the ventricle is isovolumic.7 If an exponential fall is assumed, a t1/2 (the time taken for the pressure to fall to half its initial value) can be calculated. The normal (mean (SD)) value of left ventricular t1/2 is approximately 55 (12) ms. It shortens strikingly with exercise and is prolonged with left ventricular hypertrophy or acute ischaemia. This simple exponential model may have practical limitations. Even in normal subjects, the pressure curve differs significantly from an exponential, and its asymptote is not zero. This discrepancy may be pronounced in patients with left ventricular hypertrophy or particularly with hypertrophic cardiomyopathy.
When left atrial pressure is raised, isovolumic relaxation may end prematurely so that the pressure curve has not even approximated to an exponential before filling starts. Finally, and probably most significantly, the rate of fall of pressure is profoundly affected by regional disturbances in ventricular function. It is not possible to distinguish slow relaxation from incoordinate relaxation by examining the left ventricular pressure pulse alone, and incoordination is common in disease. Nevertheless, if the entity of “impaired relaxation” which commonly figures in the literature has a defined meaning, it refers to a prolonged t1/2. The “impairment” referred to must thus be in the rate rather than the extent or timing of relaxation.
ISOVOLUMIC RELAXATION TIME
The onset of isovolumic relaxation time (IVRT), as defined by Wiggers, is aortic valve closure which can readily be determined from cusp apposition of the M mode echogram, the closure artefact on an aortic Doppler, or A2, the aortic component of the second sound on a phonocardiogram, which are all effectively synchronous. Its end is less well defined. Mitral cusp separation on the M mode precedes the onset of flow on pulsed Doppler by 10–12 ms in normal individuals and may do so by up to 100 ms in patients with disease. In normal adults the time interval from A2 to mitral cusp separation is 60 (20) ms, and that to the onset of Doppler flow approximately 85 (15) ms. Values prolong with age, so that in the elderly they are approximately double those in children. With left ventricular disease, two opposing processes can be defined (fig 1).8 The first is prolongation by the disease process itself, as seen in left ventricular hypertrophy, diabetes, or coronary artery disease. The second is shortening as left atrial pressure rises, so that with an end diastolic pressure of 30 mm Hg, isovolumic relaxation time is zero. Aortic diastolic pressure has little influence on IVRT, probably because ventricular pressure is falling rapidly at the time that the aortic valve closes. These interrelations illustrate how loading conditions interact with ventricular disease. IVRT is thus not a measure of relaxation. Normal values may arise from the combination of ventricular disease and a raised filling pressure. However, a very short IVRT is a reliable sign of a raised left atrial pressure, whereas abnormal prolongation indicates the combination of ventricular disease with normal or near normal filling pressure.

Relation between left ventricular isovolumic relaxation time (A2 to mitral cusp separation) and left ventricular end diastolic pressure in patients with coronary artery disease. Bar on horizontal axis represents the 95% confidence limits of normal. Note that isovolumic relaxation time is very short when end diastolic pressure is high. Conversely, when filling pressure is normal, isovolumic relaxation time is prolonged. This relation is unaffected by β blocker treatment.
INCOORDINATION: THE MAIN CAUSE OF PROLONGED ISOVOLUMIC RELAXATION
Although the volume of the ventricle normally remains effectively constant during isovolumic relaxation with competent valves, its shape changes towards a more spherical configuration. In addition, myocardial tagging by magnetic resonance imaging (MRI) shows rotation around the long axis and torsion between epicardial and endocardial muscle layers in normal subjects.9 Changes in cavity shape during IVRT are often accentuated in disease, particularly in association with disturbances of ventricular activation, coronary artery disease, and left ventricular hypertrophy.
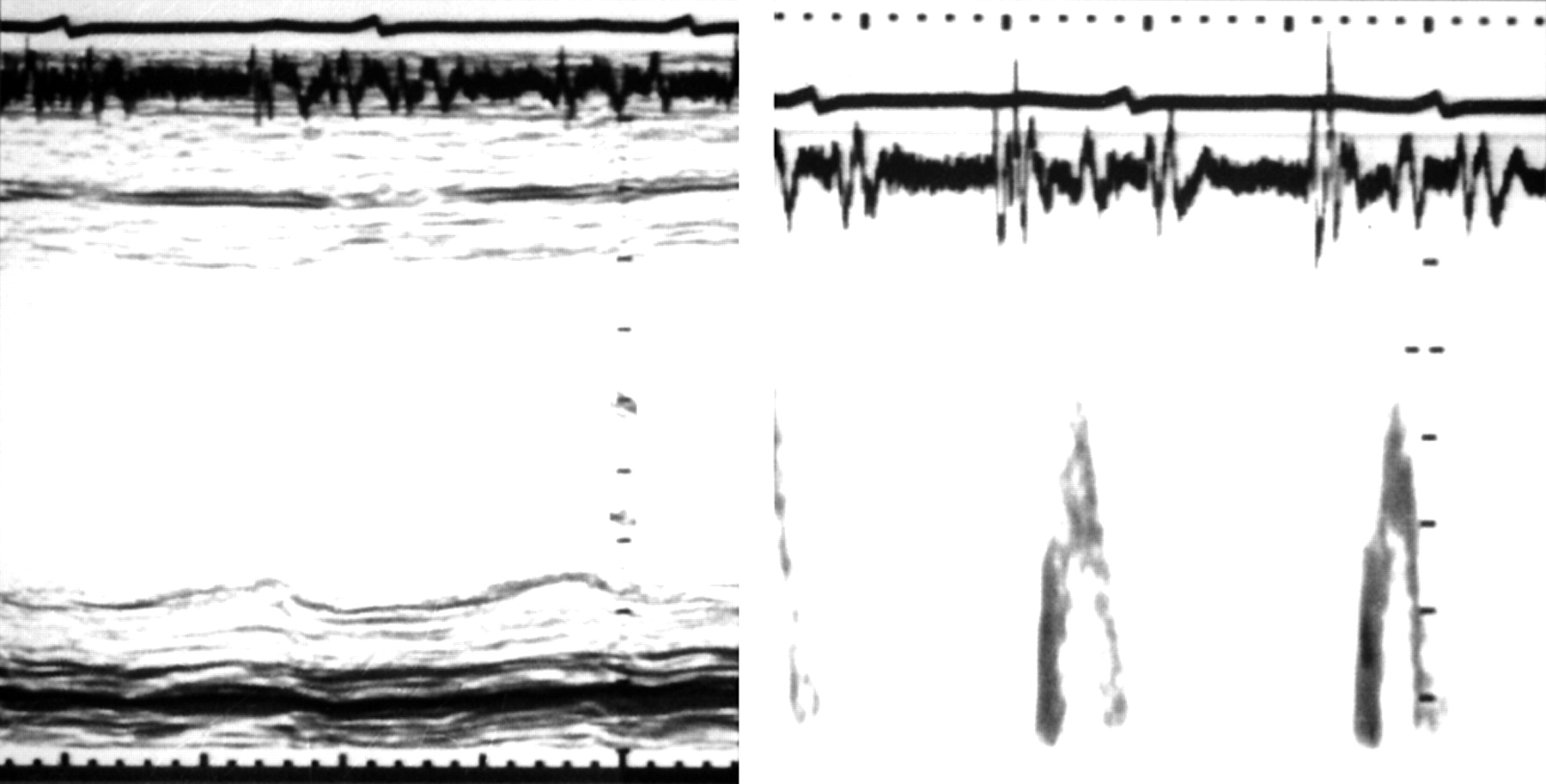
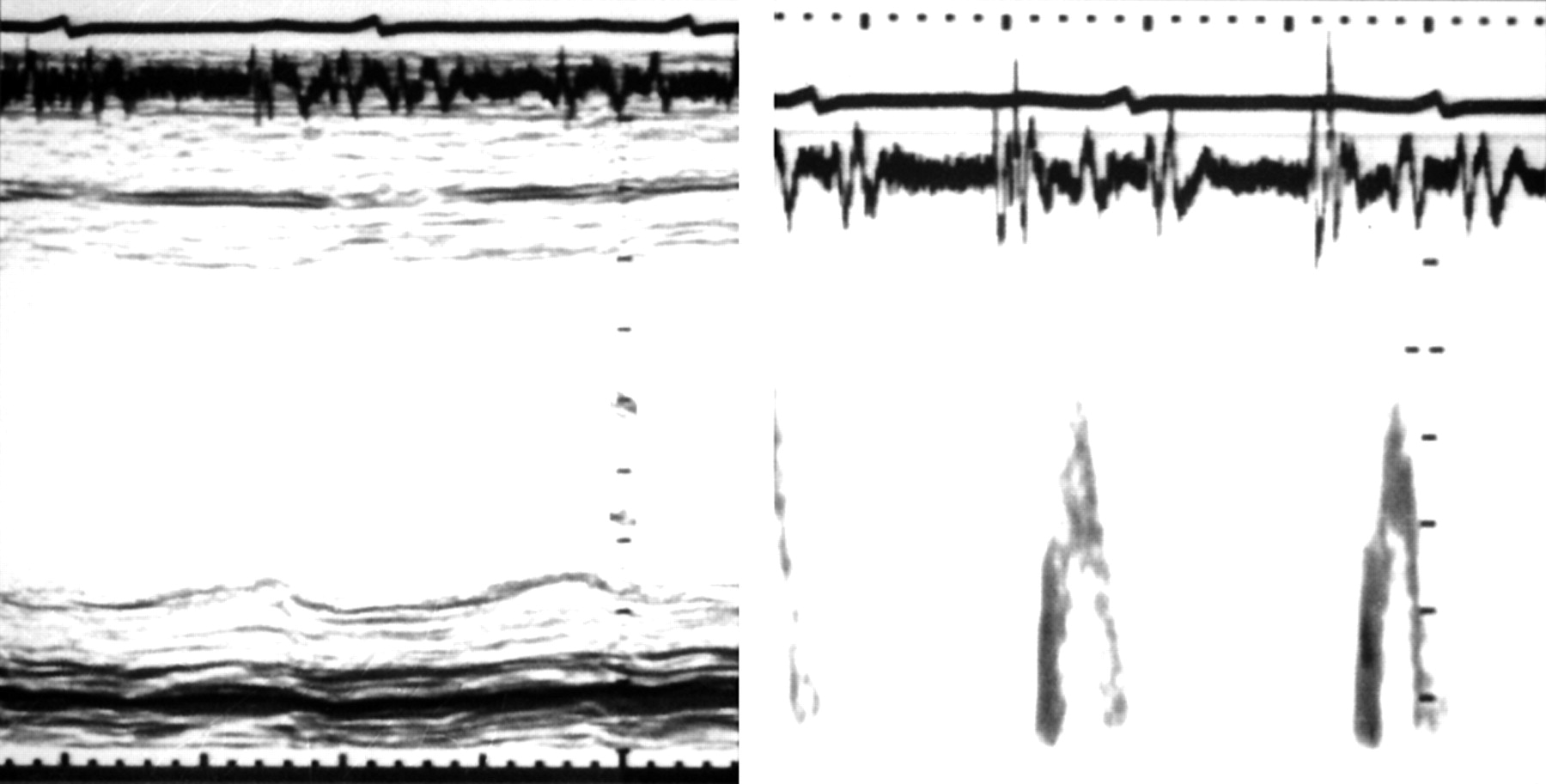
Incoordination can be usefully assessed by comparing the longitudinal motion of the left ventricle with that of the minor axis. The apex of the left ventricle has long been known to remain almost stationary throughout the cardiac cycle, so that changes in long axis can be estimated from motion of the atrioventricular ring10,11 (fig 2). This can readily be recorded by M mode echocardiography, as well as by MRI or from contrast angiograms. There are three significant components of long axis motion: amplitude, velocity, and timing. Amplitude can be measured directly and correlates closely with ejection fraction. Velocity can be derived by tissue Doppler12 (fig 3) or from digitisation of M mode records. The timing of wall motion is determined with respect to a physiological marker of overall rather than local function of the ventricle. Such a marker should be, as far as possible, insensitive to changes in heart rate or loading conditions. A2, as measured from a simple phonocardiogram, fulfils these conditions. The Q wave of the ECG is less satisfactory, since the interval from it to the onset of diastole includes left ventricular ejection, making it sensitive to changes in heart rate, stroke volume, inotropic state, and aortic pressure.13

Normal interrelations between minor (left panel) and long axis (right panel) motion. M mode echocardiograms with superimposed ECG and phonocardiograms. Note that the onset of long axis systolic motion occurs slightly earlier than on the minor axis, but that peak inward motion in both is synchronous with the second heart sound on the phonocardiogram.

Left ventricular long axis free wall motion displayed by M mode (upper trace) and tissue Doppler (lower trace), with simultaneous ECG and phonocardiogram, indicating the timing of A2 (vertical lines), from a normal subject (left) and a patient with post-ejection shortening (right). Note that in the latter patient, ventricular filling occurs almost exclusively with atrial systole. Calibration markers: upper trace, 1 cm; lower trace, 5 cm/s.
In the normal subject, circumferential and longitudinal motion are almost synchronous (fig 2). Both axes shorten throughout ejection, until A2. There is little movement during isovolumic relaxation, but during early diastole the AV ring moves rapidly upwards (that is, towards the atrial cavity) with peak velocity E', coinciding with early diastolic filling of the ventricle. It remains stationary during diastasis, and moves up again with left atrial systole (peak velocity A'). This motion of the ring around the blood was predicted many years ago on anatomical grounds.14 It contributes 10–15% of the stroke volume. This component cannot be detected by transmitral Doppler, since the blood itself remains stationary with respect to an apical transducer.
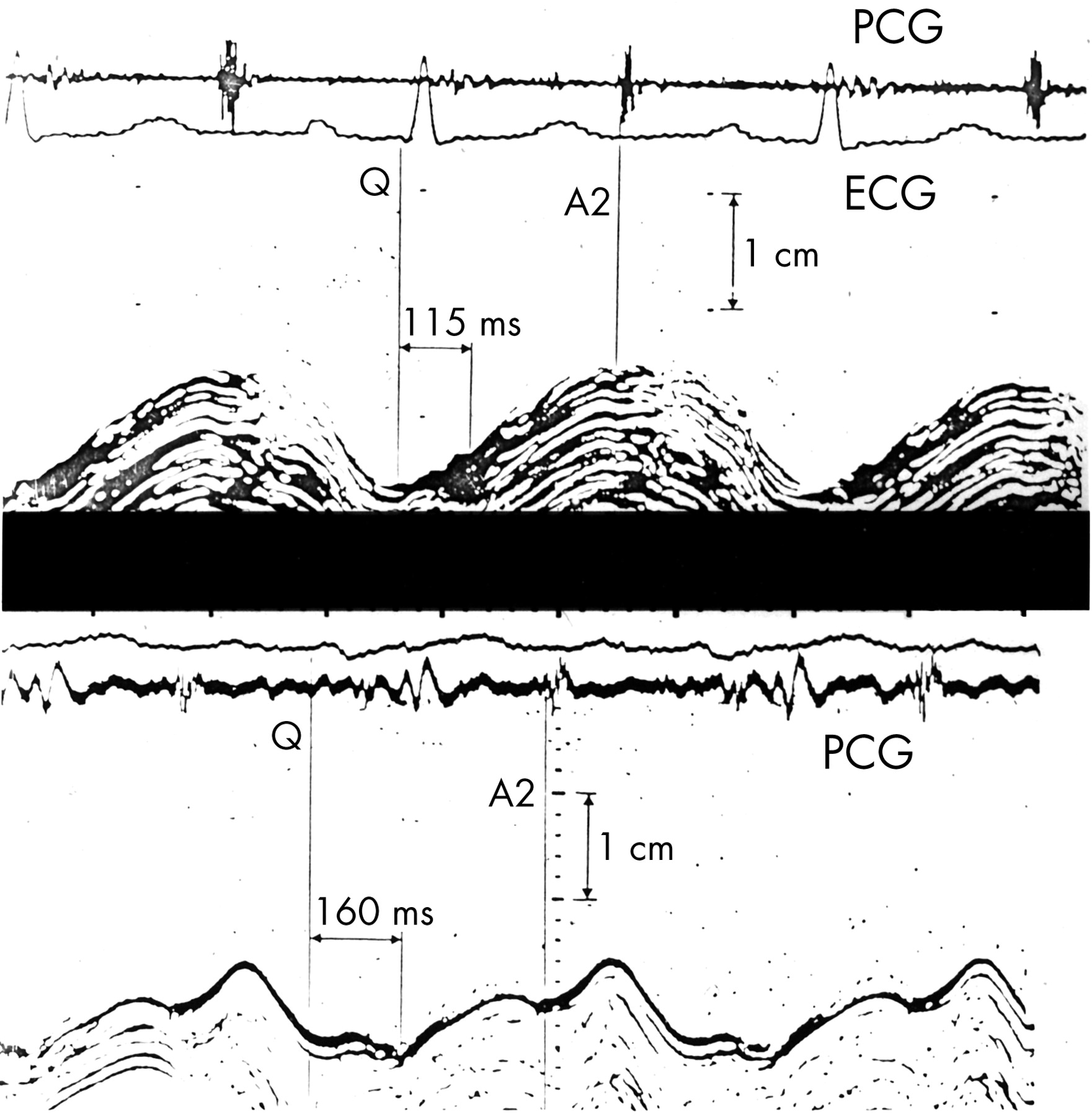
The normal synchronous pattern of AV ring motion is lost with left bundle branch block. The onset of long axis shortening is delayed with respect to the Q wave, but its overall duration is unchanged, so that tension and shortening persist well beyond the end of ejection into the period of isovolumic relaxation (fig 4).15 A similar pattern is commonly seen in patients with coronary artery disease, so that during isovolumic relaxation there is premature lengthening of the minor axis at the same time as tension persists in the long axis. Together these cause a pronounced change in cavity shape before filling starts.16 Tension development is commonly prolonged in segments supplied by stenosed coronary arteries, reverting to normal with successful intervention, either by angioplasty or vein grafting.17

Effects of intermittent left bundle branch block on left ventricular long axis motion. Top panel: with normal activation, the onset of inward motion follows that of the QRS complex by 115 ms, and that A2 (aortic valve closure) is synchronous with peak inward motion. With left bundle branch block (lower panel), the onset of inward motion is strikingly delayed to 160 ms, and peak inward motion occurs approximately 100 ms after A2.
Asynchronous relaxation has very practical consequences. It is much the most common clinical cause of a prolonged IVRT. It, rather than uniform slow relaxation, usually underlies the “impaired relaxation” invoked in disease. Blood flow must flow within the cavity of the ventricle for its shape to change.18 Such IVRT flow may show rapid acceleration and deceleration implying corresponding pressure differences within the cavity so there is no unique ventricular pressure. This casts further doubt on estimates of relaxation from rates of pressure fall. Finally, incoordinate relaxation has a major effect on the dynamics of early diastolic filling.
VENTRICULAR FILLING
Approximately 70% of the stroke volume normally enters the left ventricle during early diastole, and for the greater part of this time ventricular pressure continues to fall, although at a much slower rate than during isovolumic relaxation. The peak rate of inflow across the mitral valve at rest, measured by contrast angiography, is 500–700 ml/s. Normalised early diastolic filling rate can be determined by radionuclide angiography.19 Ventricular dimensions normally change rapidly during early diastolic filling. The transverse dimension increases, the myocardium thins, and the atrioventricular (AV) ring retracts towards the left atrium. All these processes may be slowed in patients with left ventricular hypertrophy or diabetes, even when wall motion is coordinate, and such changes have been taken as evidence of abnormal diastolic function.
The peak velocity of early diastolic velocity (in m/s) across the mitral valve can be measured by pulsed Doppler (fig 5). Values are usually in the range 1.0–1.2 m/s in young adults, falling progressively with age to 0.5 m/s or less in the elderly.20 Peak flow velocity occurs 100–150 ms after the onset of flow (the acceleration time). The corresponding velocity from the peak to the end of flow, extrapolated to the baseline if necessary, represents the deceleration time and normally has a value of 150–250 ms. The remainder of the stroke volume, normally approximately 30%, enters during atrial systole. The ratio of the two peak velocities, E/A, falls with age, so that in the elderly, atrial filling is normally dominant at rest. Measuring E/A ratio presents no difficulty when heart rate is slow, and diastasis is clearly defined. However, as heart rate increases and diastolic period shortens, the A wave becomes superimposed on the declining E wave, thus increasing its apparent amplitude. When heart rate is very fast, or the duration of diastole is reduced by abnormal prolongation of systole, E and A waves may become superimposed, leading to a single flow pulse:summation filling.

Normal transmitral flow velocities measured by pulsed Doppler echocardiography, showing early diastole (E) and atrial (A) waves. The interval from A2 on the phonocardiogram to the onset of flow (OF) represents Doppler isovolumic relaxation time. The interval from the onset of flow to peak velocity represents acceleration time (AT), and that from the peak to the end of the E wave, the deceleration time (DT).
PRESSURE GRADIENTS: THE FINAL COMMON PATHWAY
Early diastolic transmitral blood flow velocity has been directly related to a number of aspects of disease. Newton's second law of motion applies to blood as well as to all other matter. It states that the rate of change of velocity (the acceleration) is proportional to the impressed force and takes place in the direction in which the force acts. In a fluid, a force is expressed as a pressure gradient, with the physical dimensions of mm Hg/cm. (Note that the word “gradient” is often used by cardiologists to express what more correctly should be termed a pressure “difference”, which takes no account of the distance between the two points at which pressure is measured.) An acceleration of l g requires a pressure gradient of rather less than 1 mm Hg/cm. The velocity (V) of blood at any time is given by the product of acceleration (a) and the time over which that acceleration has acted (t), or V = at. Peak blood flow velocity thus depends not only on pressure gradient, but also on the time over which that gradient has acted. Conditions during filling are complex, and pressure gradient varies continuously during filling as atrial and ventricular pressure approximate to one another. This is followed by a period of pressure reversal which decelerates the incoming blood. Peak filling velocity across a normal valve thus occurs at the time when pressure gradient is zero.
Detection of a raised left atrial pressure
-
Shortening of left ventricular isovolumic relaxation time
-
Increase in peak E wave velocity and E/A ratio
-
Fall in E wave deceleration time
-
Increase in diastolic pulmonary venous velocity
-
Increase in E/E' ratio
Pressure gradient, integrated over time and space, is the only factor affecting blood flow velocity. Ventricular cavity stiffness, suction, relaxation, atrial compliance, and other external factors affect blood flow only insofar as they affect pressure gradient. Whenever blood accelerates, the appropriate pressure gradient must, by definition, be present. Unfortunately, true pressure gradients are difficult to measure in humans. They cannot be determined, for example, from atrial and ventricular micromanometers whose tips are unspecified distances apart, both horizontally and vertically. Still less can they be assessed from determinations of left atrial pressure or the pressure recorded in the ventricle at presumed mitral valve opening. Probably the most satisfactory solution is to invert the problem, and measure local blood acceleration from pulsed Doppler or MRI and derive the pressure gradients from it. Such estimates suggest that pressure gradients are strikingly increased in patients with restrictive ventricular filling.
RESTORING FORCES
The normal early diastolic pressure gradient has two components: positive left atrial pressure and a negative left ventricular pressure, referred to as ventricular suction. This suction can be demonstrated experimentally by suddenly occluding ventricular inflow at the onset of diastole, when values as low as −18 mm Hg with respect to atmospheric pressure have been recorded in normal beats and −25 mm Hg in post-ectopic beats.21 Suction arises at the end of contraction when muscle fibres have been shortened by active tension to less than an equilibrium length probably determined by their connective tissue matrix. The smaller the end systolic volume, therefore, the greater the restoring forces. More complete systolic emptying, whether as the result of vasodilatation or a positive inotropic effect, thus increases their likely effect on diastolic inflow.
Ventricular suction is difficult to quantify in humans, particularly when filling is unrestricted, since methods for apportioning atrial and ventricular components of the pressure gradient are not well developed. Furthermore, there is no general agreement as to what should be taken as the zero for pressure measurement, although it is likely that normal pericardial pressure is negative to atmospheric pressure.
The velocity of early diastolic mitral ring motion is increasingly being taken as a measure of ventricular relaxation.9 Peak early diastolic velocity correlates positively with the t1/2 of ventricular pressure fall.22 However, correlation does not necessarily indicate identity, and the two events are not synchronous. Ring motion starts at the same time as transmitral flow (fig 6)—that is, when ventricular pressure has fallen to levels below that in the left atrium. Early diastolic ring motion is synchronous with the acceleration phase of early diastolic filling, and for the latter to occur, the pressure in the left ventricle must be lower than that in the left atrium. Diastolic left ventricular pressure cannot therefore be the cause of upward ring motion. Similarly, atrial systole cannot be invoked in early diastole while ventricular relaxation is virtually complete. Thus, early diastolic ring motion probably represents release of elastic energy early in diastole from atrial and ventricular connective tissue energised during the preceding ventricular systole. In effect, it is a ventricular restoring force, whose anatomical substrate is located outside the ventricle itself. An elastic model explains the strong correlation of early diastolic velocity with total ring amplitude.

Simultaneous records of transmitral Doppler (top panel), mitral ring motion (middle), and pulmonary venous flow velocity (lowest panel). Note that the acceleration phase of transmitral flow is synchronous with early backward movement of the ring, while during ventricular ejection, forward ring motion and systolic flow from the pulmonary veins are in phase with one another.
ABNORMAL TRANSMITRAL FLOW PATTERNS
Transmitral flow velocities may be abnormal in disease and give information of clinical value. In particular, they are sensitive to left atrial pressure.23
Restrictive filling pattern
The restrictive filling pattern (fig 7)24 is recognised as a dominant E wave with a deceleration time of less than 120 ms. It is associated with a short or even zero isovolumic relaxation time and a reduced or absent A wave. Very frequently a third heart sound is present, whose onset coincides with peak of the E wave. The acceleration and deceleration rates of the E wave are both conspicuously increased, implying high pressure gradients, both forward and reversed. Reduced A wave amplitude is not usually caused by failure of left atrial contraction, since mechanical function can be demonstrated, by direct measurement of left atrial pressure, by its indirect effect on the apex cardiogram, or by detecting retrograde blood flow into the pulmonary veins. The combination of an increased atrial pressure wave with no flow across the mitral valve demonstrates increased end diastolic ventricular stiffness. Restrictive filling is good evidence of a raised left atrial pressure, which overrides any relaxation abnormality. It gives no direct information about the underlying diastolic disease. This may be specific, as occurs in amyloid or eosinophilic heart disease, or the high filling pressure may represent a complication of cavity dilatation, hypertrophy, or diabetes, or even the simple result of fluid overload distending an otherwise normal ventricle.
Records from a patient with dilated cardiomyopathy and a restrictive left ventricular filling pattern. Left panel: M mode demonstrates reduced left ventricular shortening fraction. Right panel: pulsed Doppler record demonstrates isolated E wave, although the patients is in sinus rhythm, with short acceleration and deceleration times. Note that peak transmitral velocity corresponds with a loud third heart sound on the phonocardiogram.
Dominant A wave
A value of E/A ratio lower than would be anticipated for age is commonly associated with ventricular disease.25 It is usually attributed to “impaired relaxation”, which in practice is nearly always incoordination, with long axis shortening after the end of ejection. This delayed contraction profoundly affects early diastolic filling, reducing its peak velocity or suppressing it altogether (fig 8). Clinically, this pattern is associated with the combination of ventricular disease and a low or normal filling pressure. It may thus be unmasked by a Valsalva manoeuvre. It is also common in patients initially presenting with restrictive filling who have responded favourably to treatment with diuretic and angiotensin converting enzyme (ACE) inhibitor. This sequence of events illustrates how a patient may improve clinically at the same time as diastolic measurements become more abnormal. It should not be forgotten that the most common way for diastolic measurements to normalise, or as the literature often has it, to “improve”, is for left atrial pressure to rise.

Suppression of early diastolic transmitral flow by ventricular incoordination. Normal relations between left ventricular minor axis, mitral Doppler and long axis on the left, and those from the patient on the right. Note that in the normal subject, mitral cusp separation occurs approximately 60 ms after A2, and that this is followed approximately 30 ms later by the onset of transmitral flow. In the patient, the interval A2 to mitral cusp separation is considerably prolonged, and that during this period left ventricular dimension has increased on the minor axis. The E wave of the transmitral Doppler is absent; transmitral flow occurs only with atrial systole, and its onset is delayed by 100 ms after mitral cusp separation. Long axis motion is very incoordinate, with striking shortening occurring after A2. This is maintained for nearly 200 ms, and throughout this period no transmitral flow can be detected.
“Normal” filling pattern
Just as with isovolumic relaxation time, a raised left atrial pressure and diastolic disease have opposite effects on the E/A ratio, so the combination of the two leads to a value 1.0–1.5, often referred to by the unsatisfactory term “pseudonormalisation”.26 Left ventricular filling pattern should not, however, be considered in isolation, so that recognising pseudonormality is less of a problem that the literature might suggest. The majority of patients in whom the question arises are elderly in whom an E/A ratio greater than 1 would be unusual anyway. They also have clear evidence of structural ventricular disease, either cavity dilatation or hypertrophy. In a minority, premature termination of forward atrial flow can be demonstrated, showing near restrictive filling. The ratio E/E' (peak early diastolic flow velocity to peak ring velocity) has been suggested in these circumstances. When this ratio is increased, then left atrial pressure is likely to be high. Since a low value of E' may be the result of reduced systolic amplitude, it may simply be a surrogate marker of ventricular disease. A dominant E wave in an elderly patient with ventricular disease should suggest a raised filling pressure. Recognising pseudonormality in young patients in whom a dominant E wave would in fact be normal has received little attention in the literature.
PULMONARY VENOUS FLOW
The velocity of blood flow from the pulmonary veins into the left atrium can be determined by transoesophageal or somewhat less satisfactorily by transthoracic Doppler.27 Forward flow occurs during ventricular systole (Pvs) and diastole (Pvd), and retrograde flow may occur with atrial systole (Pva). Pulmonary venous flow is divorced from right ventricular function and pulmonary artery pressure. The diastolic component is directly associated with forward flow across the mitral valve and is accentuated when ventricular filling is restrictive. The systolic component occurs well after the termination of atrial relaxation (fig 9) during the systolic or X' descent on the pressure trace. It depends on the increase in left atrial capacity caused by left ventricular long axis shortening during ventricular ejection drawing the AV ring, and thus the floor of the atrium, towards the ventricular apex. Pvs is reduced when ejection fraction, and thus ring amplitude, is low, and may thus be surrogate for left ventricular hypokinesis. As has previously been described, retrograde flow into the pulmonary veins during left atrial systole is a feature of a restrictive ventricle along with reduced ventricular systole and increased ventricular diastole.

Normal pulmonary venous flow velocities, recorded intraoperatively using transoesophageal echo in a patient with coronary artery disease, with superimposed high fidelity left atrial pressure. S, systolic wave (Pvs); D, diastolic wave (Pvd) of pulmonary venous flow; V, V wave of left atrial pressure; X and Y, X and Y descents. Note that the early diastolic wave of the pulmonary flow trace is synchronous with the Y descent of the atrial pressure wave.
Effects of age on diastolic function
-
Prolongation of isovolumic relaxation time
-
Reduction in peak E wave velocity and E/A ratio
-
Increase in E wave deceleration time
-
Fall in peak velocity of early diastolic (E') AV ring retraction.
ESTIMATING LEFT VENTRICULAR FILLING PRESSURE
A raised left atrial pressure is an important component of the clinical syndrome of heart failure, so that detecting it would be clinically useful. If ventricular filling pressure increased to the same extent as that in the atrium, filling pattern would not alter. In fact, though, a number of non-invasive approaches to estimating it in patients with left ventricular disease have proved quite effective.28 They include simple measures of isovolumic relaxation time, E/A ratio, E wave deceleration time, the ratio of systolic to diastolic pulmonary venous flow, and E/E'. All are to some extent sensitive to filling pressure, with correlation coefficients in the range 0.65–0.75. The variety of measures of filling pressure that have been used, including mean and early diastolic left atrial pressures, measured directly, by wedge or occluded pulmonary artery, as well as left ventricular end diastolic or diastolic pressure synchronous with mitral valve opening, make it difficult to compare studies. Nevertheless, the overall picture is clear: the biological range of left atrial pressure is so large that it is usually possible to distinguish a normal from a high filling pressure in individual patients with left ventricular disease by these markers. Exact values, though, are not attainable.
PASSIVE VENTRICULAR STIFFNESS
It is widely believed that ventricular stiffness is increased in disease, and that this is a common cause of the clinical syndrome of diastolic heart failure. A ventricle can only have the property of passive stiffness when pressure and volume change in the same direction. Cavity stiffness can then be defined as: change in pressure/change in volume.
Compliance is the reciprocal of stiffness.
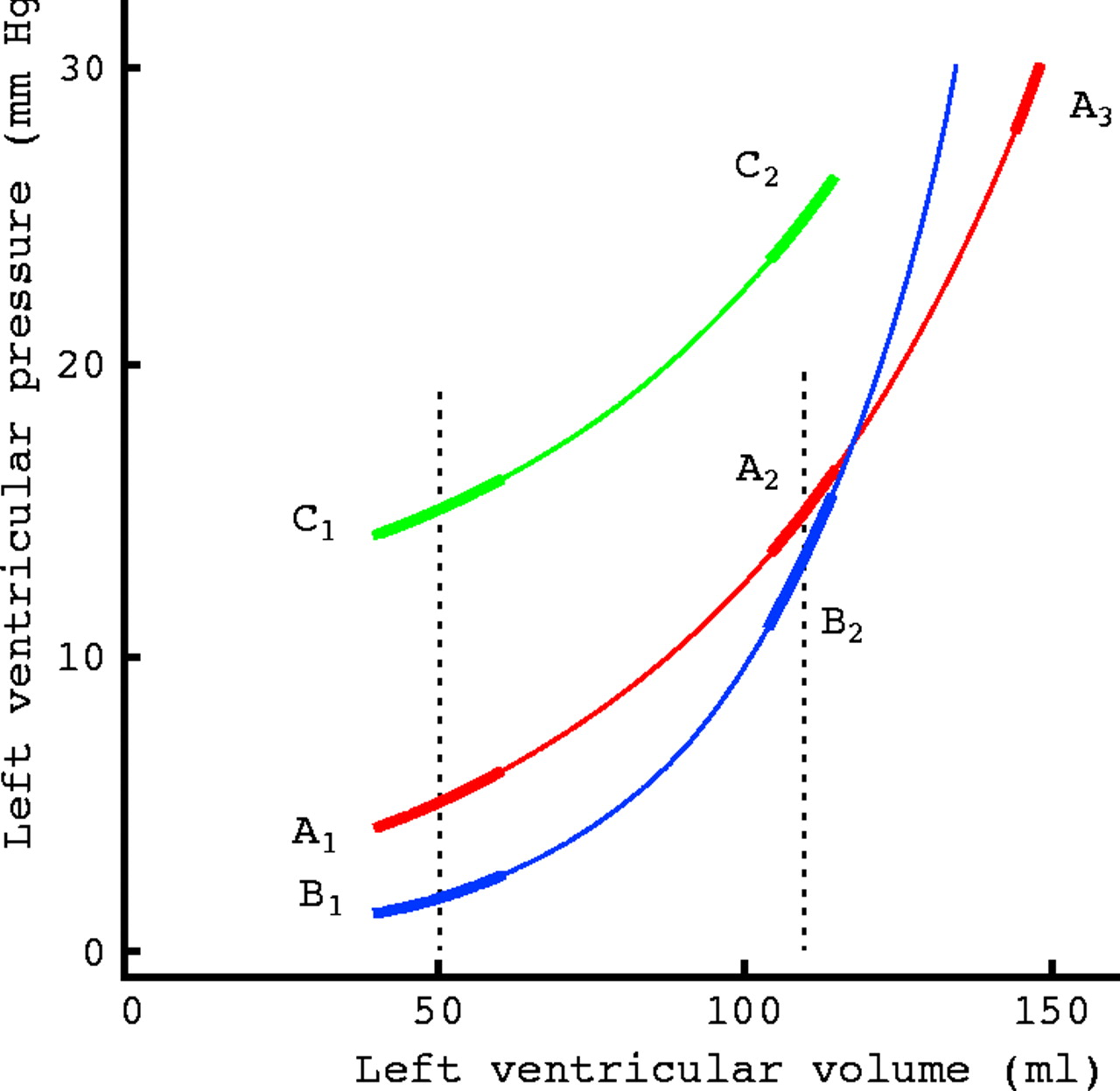
A left ventricular pressure–volume curve (A1–A3) is shown diagrammatically in fig 10. It is not linear, but its slope increases as filling proceeds. Since cavity stiffness is represented by the slope of this curve, it is clear that the cavity becomes increasingly stiff as it fills. Thus it is not possible to quote a single value for ventricular stiffness for a patient as one might for blood urea. Equally, it is not possible to devise a “standard” volume at which ventricles might be compared; a value that represents end diastole in one patient may be less than end systole in another. Since the relation between pressure and volume is curvilinear, an exponential can be superimposed without major error, and a rate or “stiffness constant” measured.29 The physical dimensions of this “stiffness constant” are, in fact, reciprocal volume (V-1) which are not those of stiffness. It cannot therefore be a measure of stiffness, but of the extent to which stiffness depends on volume—not the same thing at all.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Theoretical pressure–volume relations in humans. A1–A3 represents a normal curve and B1–B3 one with a steeper slope. Tangents to the curves—that is, ventricular stiffness—are shown. C1–C2 represents parallel upward displacement of A1–A3. See text for discussion.
The curve B1–B2 (fig 10) represents a ventricle with a higher stiffness constant, and indeed, the slope is greater at a cavity volume of 110 ml in comparison with the normal ventricle represented by A1–A2. However, if the volume of the normal ventricle is increased to 150 ml, its stiffness rises to the same high value. A normal ventricle can become very stiff if its cavity volume is increased above the physiological range as might occur with fluid overload due, for example, to renal sodium retention. Conversely, if stiffness constant is high, the cavity may become more compliant than normal when its volume is low in early diastole (B1).
Passive pressure–volume relations can become even more complex. Curve A1–A2 can become displaced to C1–C2 which is parallel to it. This change is sometimes referred as “altered distensibility”, but a better term is “parallel shift”. It characteristically occurs in an upward direction with acute angina and in a downward direction with a vasodilator. Although such an upward shift is associated with an increase in cavity pressure at any given volume, stiffness has not changed, since the slopes of A1–A2 and C1–C2 are identical. In some cases changes in right ventricular pressures and volume, possibly mediated via the pericardium, may underlie parallel shift.
Quantifying cavity stiffness has not proved very useful clinically. Not only are pressure–volume relations complex, but inconsistent values have been reported in different studies. Pressure–volume relations determined from a single beat differ from those based on reducing inflow by caval occlusion. Furthermore, the ventricle shows the property of stiffness for only a short period in mid diastole after the end of rapid filling and before the onset of atrial systole (diastasis). For diastasis to occur, heart rate must be slow, so passive stiffness relations are inapplicable in exercise. Furthermore, they represent equilibrium values, and it remains unclear whether estimates of stiffness are influenced by filling rate. In diastolic heart failure, a stiff ventricle is often invoked but seldom documented.
CONCLUSION
Although measurements made in diastole are commonly abnormal in patients with the clinical syndrome of heart failure, their contribution to understanding disturbances to intrinsic diastolic mechanisms and their management has proved modest. They are often markers of primary activation or systolic disturbances.
Paradoxically, however, their sensitivity to left atrial pressure has proved to have real clinical significance, although this is often regarded in the literature as a confounding factor. A short isovolumic relaxation time or a restrictive filling pattern indicates a raised filling pressure, a poor prognosis,30 and probably a requirement for diuretics and ACE inhibition. Incoordinate relaxation and its consequences (low or absent E wave and dominant atrial filling) are markers of disease, and their appearance in a patient in whom filling has previously been restrictive, is a sign of favourable progress.
How much an orthodox examination of diastolic function contributes to understanding the clinical syndrome of diastolic heart failure is less clear at present.31 More information is clearly needed as to the extent to which, or even whether, these abnormalities can be used to identify homogeneous patient groups, so that interrelations with symptoms and prognosis can be investigated and well designed clinical trials instituted.
REFERENCES
Linked Articles
- Miscellanea
- Miscellanea
- Miscellanea