Article Text

Statistics from Altmetric.com
Calcific aortic stenosis is the third most common cause of aortic valve disease in developed countries. This condition increases in prevalence with advancing age, afflicting 2–3% of the population by the age of 65 years.1 The aging US population has led to a burgeoning number of valve replacements per year, which in turn costs the USA approximately $1 billion. The natural history, as described by Ross and Braunwald, shows that severe symptomatic aortic stenosis is associated with a life expectancy less than five years.2 Despite the high prevalence of this condition and the increasing morbidity and mortality, very little is known regarding the cellular basis of calcific aortic stenosis.
Pathologically, progressive aortic stenosis may produce left ventricular hypertrophy, left ventricular diastolic and systolic dysfunction, congestive heart failure, angina, arrhythmias, and syncope. Recent studies demonstrate an association between atherosclerosis and its risk factors and aortic valve disease. Although a unifying hypothesis for the role of atherosclerotic risk factors in the mechanism of vascular and aortic valve disease is emerging, progress in studying the cell biology of this disease has been limited by the paucity of experimental models available. A crucial question to ask is whether the same risk factors for vascular disease that initiate an atherosclerotic type injury in the coronary arteries initiate a similar injury in the aortic valve. If this is the case, it raises the possibility that the treatments used in slowing the progression of vascular atherosclerosis may be effective in patients with aortic sclerosis. Current management of calcific aortic valve disease focuses on defining patients with valvar disease and the development of symptoms to determine the timing of surgical valve replacement. This article will review the pathogenesis, natural history, evaluation, and management of patients with calcific aortic stenosis, taking into account emerging studies important in the understanding of the cellular mechanisms of calcific aortic stenosis. It will also address diagnostic studies for the detection and diagnosis of aortic valve calcium and summarise the current standard of treatment for calcific aortic stenosis and the potential for cholesterol lowering treatment to prevent progression or aortic valve calcification.
AETIOLOGY AND PATHOGENESIS
In 1854 William Stokes described in his textbook, The diseases of the heart and the aorta, specific pathological descriptions of calcific aortic valve disease, including: (1) permanent patency of the valve in which the diameter may be increased or diminished; (2) an extreme ossific growth along the valve surrounding the ventricle, at which the valves are often destroyed; and (3) an atheromatous deposit on the ventricular surface of the valve which is often seen in the context of fatty degeneration of the heart.3 In 2003, the aetiologies of calcific aortic stenosis (table 1) include bicuspid aortic valves, degeneration, familial hypercholesterolemia, hyperuricaemia, hyperparathyroidism, Paget’s disease, ochronosis, Fabry’s disease, systemic lupus erythematosus, and drug induced valvar disease. Currently, there are many proposed theories for the cellular causation of this disease: (1) mechanical shear stress leading to calcific injury as in bicuspid aortic valves; (2) autoimmune phenomena causing degeneration; and (3) cardiovascular risk factors initiating a “response to injury” similar to that seen in atherosclerosis. Other modifying factors that are associated with an increased prevalence of aortic stenosis include conditions with a chronically raised stroke volume and altered calcium metabolism (for example, Paget’s disease, renal failure associated with arteriovenous fistula). Figure 1 shows an aortic valve removed at the time of surgical valve replacement, demonstrating valve leaflet nodule formation and thickening. These gross pathologic findings are responsible for the classic symptoms of chest pain, dyspnoea, and syncope in patients who have symptomatic severe aortic stenosis.
Aetiologies of calcific aortic stenosis, including primary valve disease and also diseases that cause secondary valve disease

Calcified human aortic valve removed at the time of valve replacement. The arrow points to the bone-like nodules on the aortic surface of the aortic valve. The leaflets also demonstrate increase in thickening.
NATURAL HISTORY
Asymptomatic patients
Patients with severe calcific aortic stenosis develop left ventricular obstruction that occurs gradually over several years. Over time the left ventricle compensates for the persistent gradient between the left ventricle and the aorta, resulting in left ventricular hypertrophy and an increase in left ventricular mass. Eventually as the aortic stenosis becomes more severe the left ventricle begins to fail. When the preload reserve is diminished and no further compensatory mechanisms are unavailable then heart failure ensues, leading to death within approximately one year.
Predictors of progression of aortic valve disease
There are a number of studies demonstrating the haemodynamic progression of this valvar lesion. Defining the rate of progression of aortic stenosis before to the onset of symptoms provides the optimal window for intervention for these patients. Doppler echocardiography has provided an accurate, non-invasive measurement of the stenosis severity, which has replaced invasive cardiac catheterisation in the majority of cases.4 The incidence of the development of symptoms in asymptomatic patients is as high as 38% in patients with Doppler outflow velocities > 4 m/s after two years and 79% after three years.5 Therefore patients with severe aortic stenosis should be monitored closely for the development of symptoms and progression of disease.
MANAGEMENT
Currently, no medical treatment is required for the asymptomatic patient with valvar aortic stenosis. Management of patients with symptomatic aortic stenosis would include selected laboratory examinations, including an ECG, chest radiograph, and an echocardiogram. The echocardiogram will confirm the presence of aortic valve disease and delineate left ventricular (LV) size and function. The American College of Cardiology and American Heart Association (ACC/AHA) guidelines have defined severe aortic stenosis as an aortic valve area of < 1.0 cm2 and a mean gradient of > 50 mm Hg.6 The decision to replace the valve is dependent on the presence or absence of symptoms with unequivocal evidence of severe stenosis, but this is a complex issue and needs to be individualised, particularly in younger active patients. Recommendations for the use of echocardiography, cardiac catheterisation, and indication for aortic valve replacement in aortic stenosis have been described in the ACC/AHA guidelines (table 2).6
Management of severe asymptomatic aortic stenosis (AS): current ACC/AHA guidelines
EMERGING CLINICAL AND EXPERIMENTAL DATA FOR THE ATHEROSCLEROTIC HYPOTHESIS
Clinical risk factors for the atherosclerosis hypothesis
Emerging epidemiological studies are revealing convincing clinical evidence towards an atherosclerotic hypothesis for the cellular mechanism of this valvar lesion. There is an increasing amount of information demonstrating the association between clinical risk factors for atherosclerosis and the development of aortic stenosis (see box). Homozygous familial hypercholesterolemia (FH) produces a particularly severe form of aortic stenosis often with supravalvar narrowing in children.7 In this specific condition extremely high low density lipoprotein cholesterol (LDL-c) concentrations are seen without the other traditional risk factors for coronary artery disease. Stewart and others from the cardiovascular health study identified several risk factors for calcific aortic valve disease: male sex, hypertension, raised LDL-c, and smoking.8 Furthermore, Palta and colleagues correlated the progression of aortic valve disease with smoking, raised serum creatinine, cholesterol, and calcium concentrations.9 These studies demonstrate that many of the same risk factors initiating vascular atherosclerosis are also implicated in aortic valve disease. Recently, Otto and colleagues reported that aortic sclerosis, which is described as focal areas of increased echogenecity and thickening of aortic valve leaflets without restricted leaflets, is found in 29% of subjects in the cardiovascular health study. In a five year follow up, aortic sclerosis was associated with an increase of approximately 50% in the risk of death from cardiovascular causes and the risk of myocardial infarction, even in the absence of haemodynamically significant obstruction to left ventricular outflow.10 These findings provide further evidence that the early lesion of aortic valve sclerosis may be associated with coronary artery disease and vascular atherosclerosis. Galante and co-workers have also demonstrated an increase in C reactive protein concentrations in patients with aortic valve stenosis.11 Finally, the extent of aortic valve calcification has been noted as an important predictor of poor outcome in patients with aortic stenosis.12 Together, these clinical studies suggest that aortic valve calcification is an inflammatory process promoted by atherosclerotic risk factors.
Risk factors for calcific aortic valve disease
-
Raised lipoprotein(a)
-
Raised low density lipoprotein cholesterol
-
Cigarette smoking
-
Hypertension
-
Male sex
-
Diabetes mellitus
-
Raised serum calcium
-
Raised serum creatinine
Histologic evidence for atherosclerosis
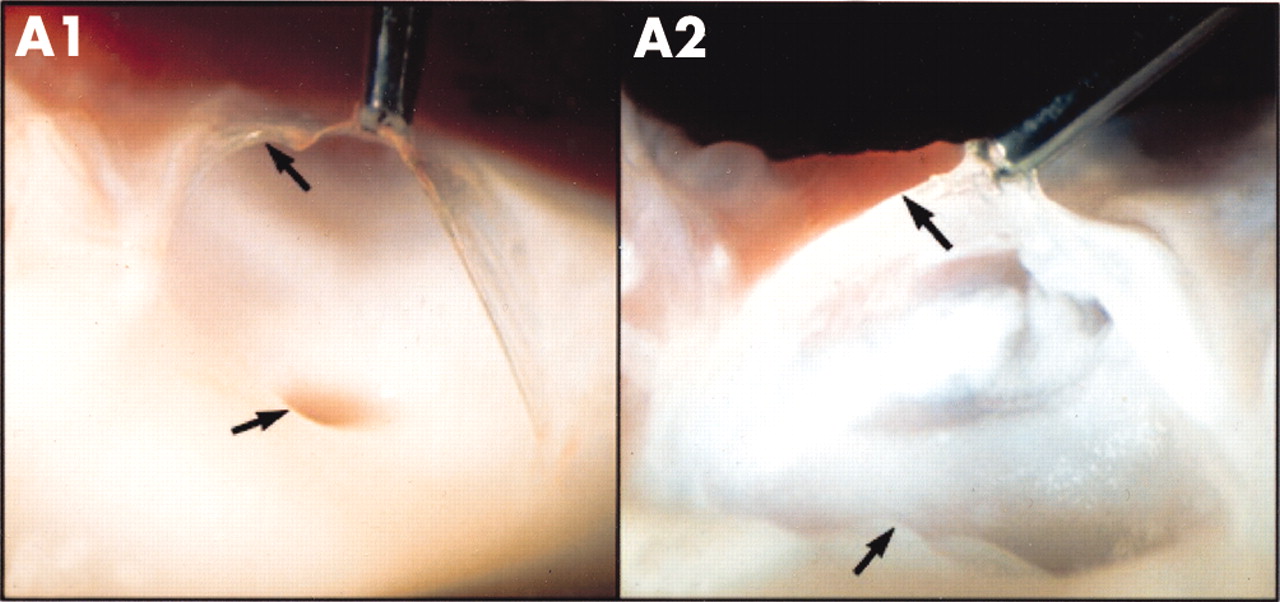
Many surgical pathological studies of human aortic valves demonstrate the presence of LDL and atherosclerosis in calcified valves, suggesting that there may be a common cellular basis for the genesis of valvar and vascular disease. O’Brien and colleagues have characterised early degenerative valve lesions in human valves and demonstrated an associated inflammatory infiltrate composed of non-foam cell and foam cell macrophages, occasional T cells, and rare α actin positive cells.13 A prominent feature of degenerative valvar aortic stenosis is the accumulation of lipid, particularly in the fibrosa, the anatomic layer of the valve located immediately below the endothelium on the aortic side of the valve.13 Co-localisation of neutral lipid with immunohistochemical staining for LDL and other proteins implicated in atherogenesis such as lipoprotein(a), and apoE containing lipoproteins might be present in aortic valvar lesions.14 These histologic studies support the hypothesis that degenerative valvar aortic stenosis is the result of an active inflammatory disease process with some similarities to atherosclerosis. These similar findings are also found in more advanced lesions but in this setting smaller amounts of α actin smooth muscles cells and more advanced calcification is present. Emerging experimental models in vitro and in vivo have demonstrated that atherosclerosis and bone-like features are also present in experimental valve calcification.15,16 Figure 2 compares a normal aortic valve with an experimental hypercholesterolaemic aortic valve in an animal model of hypercholesterolaemic diet.16 The normal aortic valve shows the presence of a normal clear glistening valve leaflet attached to the vascular aorta. The hypercholesterolaemic aortic valve shows atherosclerotic lipid deposition on the valve leaflet as well as the aorta. The combination of clinical findings and experimental observations support the hypothesis that degenerative valvar aortic stenosis is the result of an active inflammatory cellular process similar to atherosclerosis.
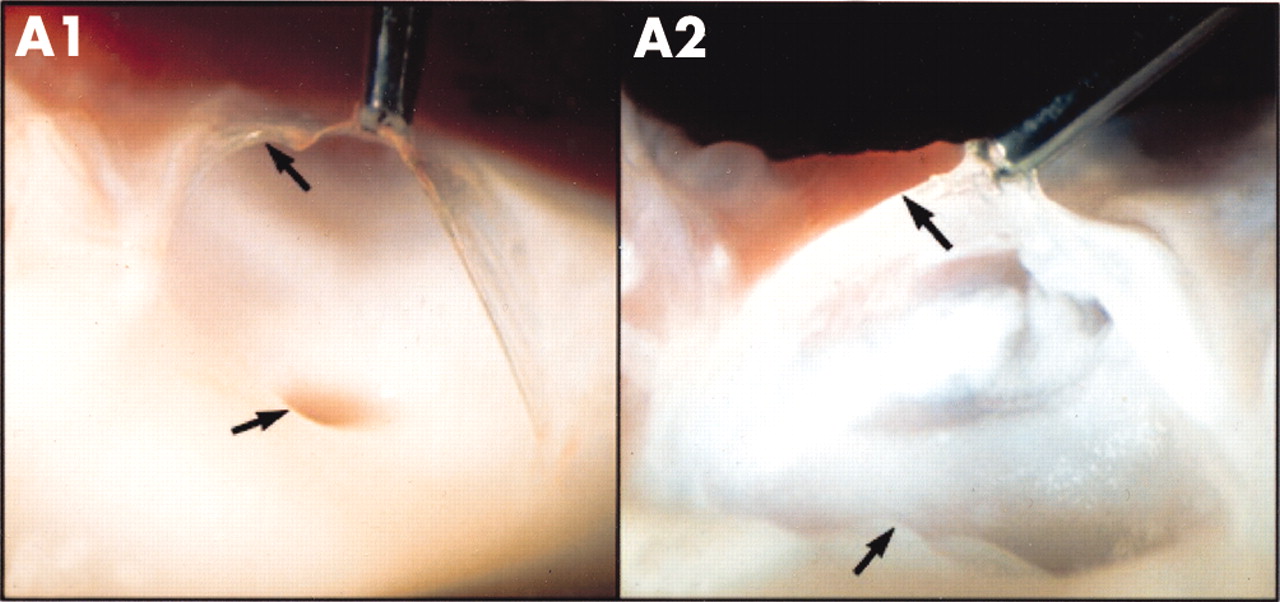
Normal versus hypercholesterolaemic aortic valve. The valve leaflet of the normal aortic valve (panel A1) has a clear glistening appearance, which is attached to the aorta. The hypercholesterolaemic aortic valve (panel A2) shows atherosclerotic lipid deposition on the valve leaflet as well as the vascular aorta.
Mechanisms of calcification
Although calcification and ossification in aortic valves has been described in the literature for over 100 years, little is known about the synthesis of bone matrix proteins in calcific aortic valve stenosis. Until recently, only descriptive histologic and protein expression studies delineating the development of calcification in the aortic valve existed. Demer and colleagues have defined cardiovascular calcification as being composed of hydroxyapatite deposited on a bone-like matrix of collagen, osteopontin, and other minor bone matrix proteins.17 Osteopontin expression has been demonstrated in the mineralisation zones of heavily calcified aortic valves obtained at necropsy and surgery.18 However, the mechanism by which this calcification occurs is not well understood. Histopathologic evidence suggests that the early lesions in aortic valves are not just a disease process secondary to aging, but an active cellular process that follows the classical “response to injury” similar to atherosclerosis.16 Comparable studies have been performed in aortic valve tissue to determine if these processes are similar. The growing body of literature suggests that hypercholesterolaemia may play a role in aortic valve calcification. Dr Rajamannan’s laboratory has studied the hypothesis that experimental hypercholesterolaemia may play a key role in the initiation and development of bone formation in valvar tissue.16 Figure 3 shows a potential mechanism for the development of aortic valve calcification. This experimental finding further substantiates the hypothesis that raised cholesterol contributes to the early injury in cardiac valves.

{kind=link}
{kind=link}
{kind=link}
Possible mechanism for the development of aortic valve calcification. The cell within the valve develops osteoblast-like features which synthesise important bone matrix proteins involved in the calcification process in the valve.
Emerging diagnostic studies for the detection and diagnosis of aortic valve calcium
Currently the “non-invasive gold standard” for diagnosis of aortic valve stenosis is two dimensional Doppler echocardiography.5 It is the test of choice to quantify the severity of valve stenosis and pressure differential across the aortic valve. However, there is growing evidence that electron beam computed tomography (EBCT) may be the most accurate non-invasive means of measuring calcification, in the coronary arteries.19 Several studies have demonstrated the utility of calculating the volume of calcium and the rate of progression of the disease process in the aortic valve.20 These are still preliminary studies which require validation in larger numbers of patients. The utilisation of EBCT may prove more sensitive in the accuracy of measuring the volume of calcification in the valve.
FUTURE MEDICAL TREATMENTS
The natural history studies of valvar aortic stenosis, as defined by clinical and histopathologic parameters, have provided landmark developments towards the understanding of this disease. Hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) may allow innovative therapeutic approaches employing both lipid lowering and possibly non-lipid lowering effects to forestall critical stenosis in the aortic valve. These agents have potent LDL lowering effects via inhibition of the rate limiting step in cholesterol synthesis. The evidence to date suggested that three putative non-lipid lowering mechanisms may contribute to the beneficial effects of HMG-CoA reductase inhibitors in coronary artery disease. These include modification of (1) endothelial function, (2) inflammatory responses, and (3) thrombus formation. Valve replacement is, and will remain, the treatment of choice for severe critical aortic stenosis. Future insights into the mechanisms of calcification and its progression may indicate a role for lipid lowering treatment in halting or modifying the rate of progression of stenosis, and the potential for such an approach is intriguing. Circumstantial, preliminary evidence suggests that aggressive treatment with lipid lowering agents may have a positive impact on the progression of valvar calcification. Novaro and colleagues have published a retrospective study showing less progression of aortic stenosis in patients on statin treatment as assessed by a decrease in the peak and mean gradient of the treated patients.21 Shavelle and colleagues also evaluated the effects of statin treatment on the amount of calcium present as measured by EBCT; retrospective analysis showed that statin treatment was associated with a 62–63% lower rate of aortic valve calcium accumulation as assessed by percentage change per year in the scoring system developed for the assessment of the amount of calcium present in the cardiovascular system.22 Table 3 summarises the retrospective data for the use of statins in the treatment of calcific aortic valve stenosis.23,24 These retrospective studies, combined with the experimental data, suggest that statin treatment may have a role in the inhibition of bone matrix production and end stage calcification, as proven in the regression of coronary artery calcification.
Retrospective studies of statin treatment in calcific aortic valve stenosis
SUMMARY
This new evidence agrees with the original descriptions of Dr Stokes in that there is an atherosclerotic ossified lesion contributing to the pathogenesis of this disease.3 Recent epidemiological clinical studies have revealed that the risk factors for arterial atherosclerosis—male sex, smoking, and raised serum cholesterol—are similar to the risk factors associated with development of aortic valve stenosis.8 Experimental studies in vitro and in vivo support the hypothesis that the development of aortic valve calcification is similar to that of vascular calcification.15,16 Preliminary data are also emerging towards the potential possibility of primary and/or secondary prevention of aortic stenosis. Moreover, future clinical studies using statins in the treatment of aortic valve disease are also necessary to determine their potential for slowing this disease process in patients.
REFERENCES
Linked Articles
- Miscellanea
- Miscellanea