Article Text

Statistics from Altmetric.com
- GUSTO IV-ACS, global use of strategies to open occluded coronary arteries IV – acute coronary syndromes
- PCI, percutaneous coronary intervention
- SVG, saphenous vein graft
Coronary atherosclerosis is the underlying cause of nearly all cases of ischaemic heart disease, and superimposed thrombosis is the cause of the great majority of acute coronary syndromes.1,2 The pathogenesis of peripheral arterial disease and, to a great extent, ischaemic stroke is similar. Thus, “atherothrombosis” is the leading cause of severe disability and cardiovascular death.
ATHEROTHROMBOTIC BURDEN
In general, atherothrombotic plaques responsible for acute coronary syndromes are larger (hidden in positively remodelled arteries) and softer (contain more lipid, inflammation and thrombus and less calcification) than angina producing lesions.1,2 Plaques in aortocoronary saphenous vein grafts (SVG) are, in general, extraordinarily bulky, friable, and thrombus-rich, regardless of clinical presentation.3 Recent observations indicate that the atherothrombotic burden is a major determinant of coronary microembolisation, particularly when plaques are crushed and fragmented mechanically during percutaneous coronary interventions (PCI).
Saphenous vein graft lesions
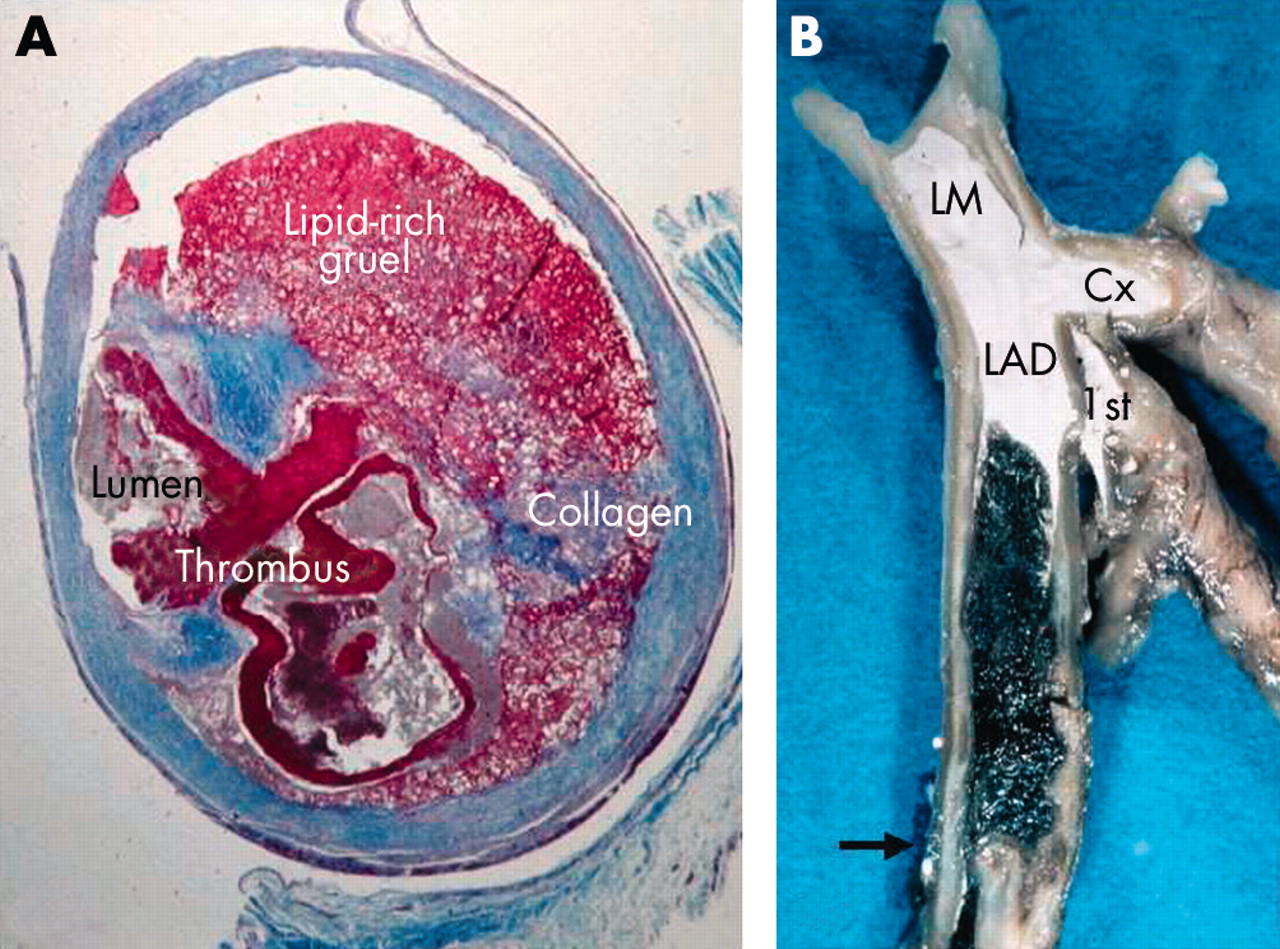
Atherogenesis is notably accelerated in SVGs, and fatal atherothrombosis may develop within a few years after grafting (fig 1A).3,4 Compared to atherothrombosis in native coronary arteries, plaques in SVGs are generally much larger and contain more lipid, inflammation (foam cells), and thrombus and less calcification.3 Consequently, the atherothrombotic burden is larger and the plaques are much more friable (vulnerable), which explains the exceptionally high risk of distal embolisation, particularly when old SVGs are manipulated by surgeons’ hands or cardiologists’ devices.
{kind=link}
Coronary microembolisation and no reflow is particularly common in two clinical settings; old stenotic saphenous vein grafts (A, high plaque burden) and acute coronary occlusion (B, high thrombotic burden). Where does all this atherothrombotic material go during percutaneous coronary intervention? (A) Cross-section of a four year old saphenous vein graft, revealing a bulky and lipid-rich (friable) plaque with ruptured surface and superimposed thrombosis (trichrome stain). (B) Left coronary artery opened longitudinally, revealing a dark red stagnation thrombus (erythrocyte-rich) between the first diagonal branch (1st) and the occlusive white thrombus (platelet-rich) formed on top of a ruptured plaque (arrow). White contrast medium injected postmortem is filling the lumen proximal to the thrombus. LM, left main; LAD, left anterior descending artery; Cx, circumflex artery.
Coronary thrombosis
Approximately 75% of coronary thrombi are precipitated by rupture of a soft and “vulnerable” plaque,1,2 and the same mechanism underlies atherothrombotic occlusion of SVGs.4 Platelet aggregation plays a critical role initially during the evolution of a coronary thrombus, but blood stagnation and coagulation contribute significantly to the overall thrombotic burden once the platelet-rich thrombus occludes the lumen totally (fig 1B). Lack of side branches favour blood stagnation and an enormous amount of thrombus may develop in occluded large calibre SVGs. Furthermore, thrombi in SVGs often persist and organise slowly, if at all.
ATHEROTHROMBOTIC MICROEMBOLISATION
Lipid-rich and inflamed plaques are vulnerable to rupture. By rupturing, the soft atheromatous gruel is suddenly exposed to the flowing blood which increases the risk of both local thrombosis and distal embolisation of atherothrombotic material.5,6 The latter phenomenon, known as athero- or cholesterol embolisation, has been described in aorta, carotid, coronary, and other arteries. Superimposed thrombosis may seal a ruptured plaque and prevent distal atheroembolisation, but thrombolysis may re-expose the soft gruel and thus revive the risk of atheroembolisation.
Spontaneous coronary microembolisation
A ruptured plaque with a superimposed non-occlusive thrombosis can, in principle, shower and obstruct the microcirculation with soft plaque material (atheroembolisation) and/or thrombotic material (thromboembolisation). Postmortem studies of patients who died after a thrombus mediated heart attack have revealed thromboemboli and, more rarely, atheroemboli impacted downstream in small intramyocardial arteries in a high proportion of cases.6–9 The overall microembolic burden is unknown but is probably relatively low compared to what may happen after PCI. Troponin elevations in acute coronary syndromes without ST elevation indicate (micro)infarction of thrombotic, but not necessarily thromboembolic, origin.5 A dynamic atherothrombotic lesion in an epicardial artery may cause subendocardial ischaemia and (micro) necrosis by reducing the blood flow subcritically and/or temporarily without implicating microembolisation. The marginal, if any, protective effect of platelet glycoprotein IIb/IIIa inhibition in acute coronary syndromes without ST elevation treated conservatively (no PCI) could indicate that spontaneous platelet mediated microembolisation plays no major role in this syndrome.10 Even troponin positive patients did not receive any benefit from potent platelet inhibition in the large GUSTO IV-ACS trial.10
Iatrogenic coronary microembolisation
In myocardial infarction with ST elevation, fibrinolytic treatment alone is capable of promoting distal embolisation, but mechanical crushing and fragmentation of the culprit lesion during PCI has emerged as the major cause of coronary microembolisation.5,6 The risk of PCI mediated microembolisation depends on the atherothrombotic burden and the invasiveness of the procedure.11 Consequently, it is relatively common in two clinical settings: PCI in stenotic SVGs (bulky and friable plaques, fig 1A) and in acute myocardial infarction (soft plaques + thrombosis, fig 1B); atherectomy and stenting cause more plaque fragmentation and distal embolisation than balloon angioplasty.11 Thus, despite otherwise successful recanalisation, PCI induced distal microembolisation and microvascular obstruction may lead to inadequate myocardial perfusion, the so-called “no reflow” phenomenon.
NO/SLOW REFLOW
The pathogenesis of no/slow reflow after PCI in atherothrombotic heart disease differs significantly from the “classical” no reflow phenomenon seen after temporary occlusion of normal coronary arteries in animals.12
Classical no reflow in animals
After coronary occlusion in dogs, myocytes begin to die in the subendocardial myocardium after ~20 minutes, and ischaemic cell death progresses from the subendocardium to the subepicardium as a wavefront in a time dependent fashion.12 About six hours of ischaemia are required to complete the wavefront of necrosis. Although myocardial necrosis averaging 28% of the vascular bed has developed after 40 minutes of ischaemia, myocardial reperfusion is still homogeneous without any defect if coronary flow is restored. However, if coronary flow is not restored until after 90 minutes of ischaemia, myocardial perfusion defects (no reflow) are now present in myocardium that was necrotic at an earlier time point, first in the subendocardial zone.12 No reflow, or more correct “no reperfusion”, appears to be confined to myocardium that is already necrotic and thus follows necrosis and not vice versa. To date, no study of “pure” coronary occlusion (that is, without microembolisation) have demonstrated myocardial no reperfusion preceding myocardial necrosis.12 The no reperfusion areas enlarge both with the degree and duration of ischaemia and with the duration of reperfusion (combined ischaemia and reperfusion injury).12,13
The inability to reperfuse necrotic myocardium is caused by progressive microvascular occlusion. Many different obstructive mechanisms have been proposed such as endothelial swelling, neutrophil plugging, vascular “squeezing” by ischaemic contracture (intracellular calcium overload), or compression from the surrounding necrotic and swollen myocytes.12 Microvessels plugged by platelets and fibrin are also seen, but microembolisation does not occur and fibrinolytic treatment is ineffective in this model.
No reflow after PCI in myocardial infarction
Unlike animal models of coronary occlusion, the clinical setting involves an atherothrombotic occlusion with its innate risk of distal embolisation when crushed or fragmented mechanically. Thus, coronary no/slow reflow and myocardial hypoperfusion after otherwise successful recanalisation of infarct related arteries may involve more than just classical no reperfusion confined to myocardium that is already dead. No/slow reflow may also result from PCI induced microvascular obstruction caused by distal microembolisation and/or microvascular spasm.5,6 Because microemboli necessarily stream preferentially to well perfused and viable myocardium, microembolisation kills potentially salvageable myocardium. Thus, the vital question is, of course: how much of the coronary no/slow reflow and myocardial hypoperfusion seen after primary PCI reflects the classical no reflow phenomenon caused by necrosis, and how much reflects PCI induced distal microembolisation (and microvascular spasm?) causing more necrosis? The (athero)thrombotic burden may prove to be critical, indicated by the beneficial effect of platelet glycoprotein IIb/IIIa receptor inhibition before stenting.14 Whether thrombectomy (before PCI) or distal embolic protection devices (during PCI) will improve myocardial perfusion and clinical outcomes remains to be shown by ongoing clinical trials.
No reflow after PCI in old SVGs
PCI of stenotic SVGs is associated with an exceptionally high risk of macroembolisation (angiographic distal cutoff) and no or low flow through myocardium that was perfused normally before PCI.15 PCI induced distal microembolisation and/or microvascular spasm are the most obvious explanations (classical no reflow is irrelevant), recently documented by the pronounced reduction in procedure related no reflow and myocardial infarction with the use of a distal embolic protection device during stenting.15 The lack of a consistent benefit with the use of potent antiplatelet agents during PCI in SVGs indicates that embolised atheromatous debris rather than platelet mediated thromboembolism is responsible for the detrimental effects associated with stenting of stenotic SVG lesions.15,16