Article Text

Statistics from Altmetric.com
Chronic heart failure (CHF) represents a major public health burden, and its prognosis is comparable to that of different malignant diseases. Our understanding of CHF has developed from the rather simplistic model of mere pump failure to that of a multisystem disorder which affects not only the cardiovascular system but also the musculoskeletal, renal, neuroendocrine, and immune systems. Thus, the pathophysiology of CHF is exceedingly complex. Therapies to block excessive neuroendocrine activation have become a cornerstone of treatment. CHF progresses because of activation of neurohormones and pro-inflammatory cytokines following an initial cardiac injury or a mutation of the genetic programme.1 Virtually any heart disease can ultimately lead to heart failure, although the initial event leading to the development of this syndrome is in many cases unknown. However, CHF is always the result of some underlying process and the diagnosis cannot stand alone.
The activation of the aforementioned systems maintains and worsens CHF. In particular, the activation of the immune system has received considerable interest in the last decade. There are several different components to this system which interact with each other in a complex manner. It is becoming increasingly apparent that inflammatory mediators play a crucial role in the development of CHF, and several strategies to counterbalance different aspects of the inflammatory response are considered. Possible targets involve pro- and anti-inflammatory cytokines and their receptors, endotoxin, adhesion molecules, nitric oxide and nitric oxide synthase, reactive oxygen species, and different types of leucocytes. The purpose of this review is to give a brief overview of the current understanding of the role of inflammation in CHF. Furthermore, we will discuss recent advances in the development of new therapeutic strategies in this field.
ROLE OF PRO-INFLAMMATORY CYTOKINES
Cytokines form a vast array of relatively low molecular weight, pharmacologically active proteins. These substances are secreted by different cell types for the purpose of altering either their own function (autocrine) or that of adjacent cells (paracrine). The most important cytokines implicated in the progression of CHF are tumour necrosis factor α (TNFα), interleukin (IL) 1, and IL-6. These cytokines share some of their major characteristics (redundancy), and all act in a pro-inflammatory sense. Among those, TNFα is the cytokine which has been studied in greatest detail.
Several hypotheses have been suggested to describe the origin of immune activation in CHF (fig 1⇓). The production of pro-inflammatory cytokines has mostly been attributed to secretion by mononuclear cells, although the myocardium seems to be another important source. Some evidence suggests that catecholamines augment this myocardial cytokine production. The concepts trying to explain increased production of pro-inflammatory mediators comprise response to myocardial injury2 and underperfusion of peripheral tissues3 (fig 1⇓). We have proposed that increased bowel wall oedema causes translocation of bacterial endotoxin from the gut which eventually yields pro-inflammatory cytokine production from monocytes in the bloodstream and possibly other tissues.4 Indeed, endotoxin, also known as lipopolysaccharide (LPS), is one of the strongest inducers of TNFα and other pro-inflammatory mediators (fig 2A⇓). Very small amounts of this substance are capable of inducing TNFα secretion.5 Attempts to prove any of the aforementioned hypotheses, however, yielded only indirect evidence. Thus, the elevation of the plasma concentrations of several pro-inflammatory mediators in acute myocarditis and acute myocardial infarction may hint towards the tissue injury hypothesis.2 Also, peripheral IL-6 spillover was found to be increased in patients with CHF when comparing arterial and venous plasma concentrations, thus indicating a peripheral cytokine production.3 This, however, has not been demonstrated for TNFα. Moreover, soluble CD14, a marker of endotoxin–cell interaction and shedding from the cell membrane (fig 2A⇓), was found to be increased in some patients with CHF (controls: 2714 (121) ng/ml; CHF patients: 3401 (134) ng/ml, p = 0.0048), especially in those with cachexia.4 It was subsequently shown that LPS concentrations are raised in CHF patients with peripheral oedema and that diuretic treatment results in reduction of LPS values.6 It might eventually be found that all of the above mechanisms take part in what is now known as immune activation in CHF.

Competing theories. Several theories to explain the cause(s) of inflammatory immune activation in CHF have been suggested. These theories may in fact complement each other. The myocardium itself is able to release pro-inflammatory cytokines, which is augmented by adrenergic stimulation. Myocardial tissue injury—for example, myocardial infarction, bacterial translocation, and peripheral tissue hypoxia—may lead to mononuclear cell activation, which eventually leads to pro-inflammatory cytokine activation. See text for details. LPS, lipopolysaccharide; LV, left ventricle; RV, right ventricle.
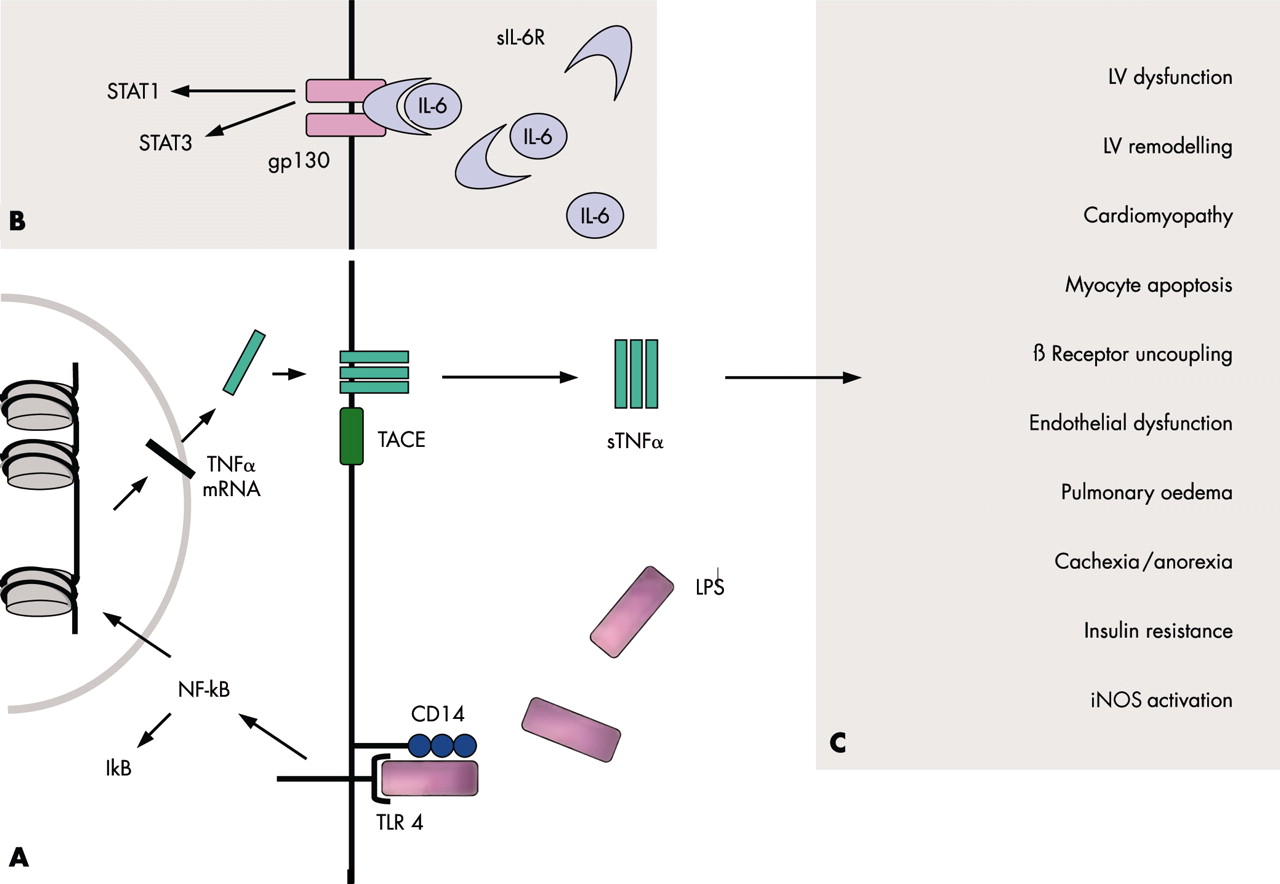
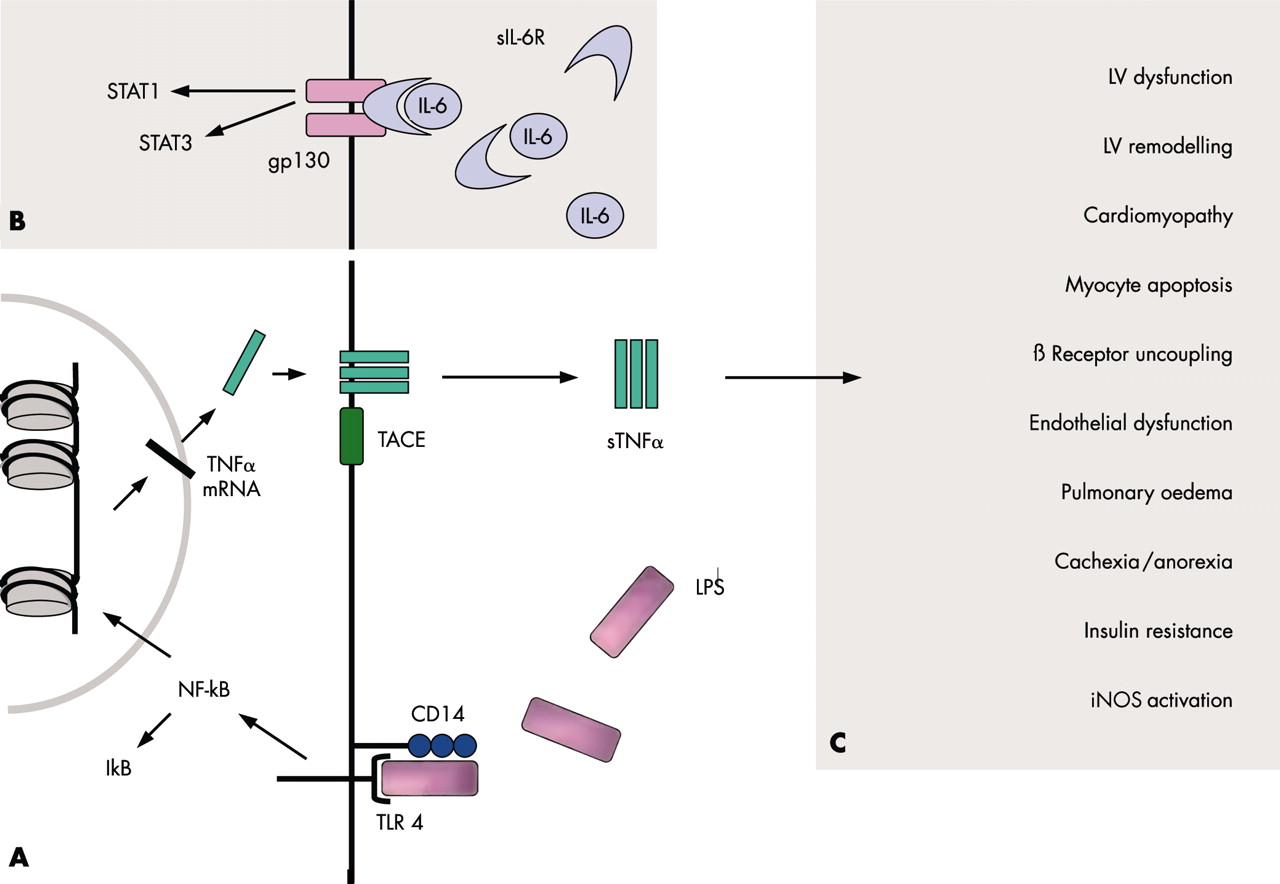
(A) TNFα signalling via a receptor complex comprising CD14 and toll-like receptor 4 (TLR4). Upon LPS binding to this complex, signal transduction via TLR4 leads to the activation of the transcription factor NF-κB. Therefore, IκB is cleared off the molecule. After transcription and translation, trimeric TNFα is inserted into the cell membrane. The TNFα converting enzyme (TACE) cleaves TNFα off the membrane to yield its soluble form (sTNFα). (B) IL-6 signalling via IL-6R and gp130. Only the latter has to be expressed to render the respective cell susceptible to IL-6, because soluble IL-6R can interact with membrane bound gp130. The activation of the receptor complex leads to the activation of different transcription factors, such as STAT1 and STAT3. (C) TNFα mediated effects. Several untoward effects have been implicated into the over-expression of TNFα. See text for details. gp, glycoprotein; IκB, inhibitory κB; IL, interleukin; iNOS, inducible isoform of nitric oxide synthase; LPS, lipopolysaccharide; LV, left ventricular; NF-κB, nuclear factor-κB; R, receptor; s, soluble; STAT, signal transducers and activator of transcription; TACE, TNFα converting enzyme; TNFα, tumour necrosis factor α.
Tumour necrosis factor α
TNFa was first described in 1975 and termed cachectin. In 1990 Levine and associates observed that mean (SEM) serum concentrations of TNFα were higher in CHF patients than in healthy subjects (115 (25) v 9 (3) U/ml, p, 0.001).7 They also demonstrated that those patients with high concentrations of TNFα were more often suffering from cardiac cachexia.7
TNFα exerts its effects via TNFα receptors (TNFR), which are expressed by almost all nucleated cells. Two TNFRs have so far been identified.8,9 TNFR-1 is more abundantly expressed and appears to be the main signalling receptor. The majority of deleterious effects caused by TNFα seem to be mediated via this receptor, whereas TNFR-2 appears to have a more protective role in the heart. Previous studies have identified both types of receptors in non-failing and failing human myocardium. After translation, both receptors, like TNFα itself, are inserted into the cell membrane of the respective cell.8,9 Proteolytic cleavage by TNFα converting enzyme (TACE) yields the soluble forms (fig 2A⇑). The precise role of the soluble TNFRs remains unclear, although some evidence suggests that they stabilise the TNFα molecule, thus potentiating its detrimental long term actions. However, higher concentrations of TNFRs appear to inhibit TNFα activity. It is thought that high plasma concentrations of soluble TNFRs primarily indicate a history of raised TNFα values. The reproducibility of plasma concentrations of soluble TNFRs is higher than that of TNFα itself. This may be the reason why soluble TNFRs predict short term10 and long term11 prognosis better than TNFα in CHF patients.
Several untoward effects seem to be associated with raised TNFα production in CHF (fig 2C⇑). TNFα has been implicated in the development of left ventricular dysfunction, left ventricular remodelling, increased cardiac myocyte apoptosis, the development of anorexia and cachexia, reduced skeletal muscle blood flow and endothelial dysfunction, severity of insulin resistance, activation of the inducible form of nitric oxide synthase (iNOS), β receptor uncoupling from adenylate cyclase, and other effects.1,8,9
Interleukin 6
TNFα is not the only cytokine which worsens CHF. According to the cytokine hypothesis, CHF progresses because cytokines exacerbate haemodynamic abnormalities or exert direct toxic effects on the heart.12 Increased concentrations of IL-6 have been shown in the circulation of CHF patients. IL-6 can produce myocyte hypertrophy, myocardial dysfunction, and muscle wasting. On the other hand, IL-6 seems to block cardiac myocyte apoptosis. Since IL-6 is thought to be released in direct response to TNFα, it was not surprising that a linear correlation between the two was observed. Therefore, the existence of a cytokine cascade has been suggested.12
While increased concentrations of IL-6 were found to be associated with a poorer prognosis in CHF patients, those of the soluble IL-6 receptor (IL-6R) were not. Interestingly, it is a small transmembrane glycoprotein termed gp130, but not IL-6R itself, which renders cells susceptible to IL-6 (fig 2B⇑). Indeed, IL-6 can act on cells lacking the expression of IL-6R after complex formation with soluble IL-6R. Both gp130 and IL-6R are always required for signalling. The soluble form of gp130 inactivates the soluble IL-6/IL-6R complex. However, both the concentrations of gp130 and the overall level of bioactivity of IL-6 are increased in CHF.
Interleukin 1
Another important cytokine in the setting of CHF is IL-1. IL-1 along with TNFα are generally thought of as prototypical pro-inflammatory cytokines. IL-1 has been demonstrated in the myocardium of patients with idiopathic dilated cardiomyopathy, and it depresses myocardial contractility in a dose dependent fashion. This effect is synergistic with that of TNFα, and the stimulation of iNOS seems to be involved. Additional findings have shown IL-1 being involved in myocardial apoptosis, hypertrophy, and arrhythmogenesis.
When summarising the role of IL-6 and IL-1 in the setting of CHF one must admit that the precise mechanisms involved are far from being entirely clear. In fact, both substances also exert some beneficial (cardioprotective) effects, although the role of these remains to be elucidated in more detail.
Downstream signalling pathways
Nuclear factor κB (NF-κB) is crucially involved in the regulation of a battery of inflammatory genes, including pro-inflammatory cytokines, chemokines, and adhesion molecules. The NF-κB family consists of a number of proteins, which make up homo- and heterodimers to form the active transcription factor. In unstimulated cells, cytoplasmic NF-κB is bound to its inhibitory protein IκB with its nuclear localisation signal being masked (fig 2B⇑). Different stimuli, among them a good many of those that are regulated by NF-κB itself, lead to IκB degradation. This leads to NF-κB translocation into the nucleus, where it binds to promotor or enhancer regions of specific genes.
Only very few data about NF-κB in CHF are available. Since pro-inflammatory cytokines are raised in CHF, it is tempting to speculate that NF-κB activity is elevated in the respective cells as well. Indeed, myocardial tissue from patients with CHF exhibits activation of NF-κB. A study in 16 patients with end stage heart failure showed that left ventricular assist devices, used to bridge the time until transplantation, reduced the number of NF-κB positive cardiomyocyte nuclei significantly.13 Healthy hearts do not normally express NF-κB to a detectable extent. In the light of these findings it has been speculated that NF-κB is involved in what is called “reverse remodelling” of the heart during treatment with such devices.
Interleukin 10
IL-10 is one of the most important anti-inflammatory cytokines. It is known to down regulate the production of TNFα, IL-1, and IL-6, respectively. This has been confirmed in LPS stimulated peripheral blood mononuclear cells from CHF patients.14 Furthermore, IL-10 limits the production of macrophage derived nitric oxide (NO) and oxygen-free radicals. IL-10 also enhances the release of soluble TNFR, which contributes to the reduction of TNFα activity. Similar to pro-inflammatory cytokines, IL-10 mRNA has also been detected in the failing myocardium. Circulating IL-10 concentrations have been reported to be either increased or decreased in CHF patients compared to healthy, age matched controls. However, the intravenous administration of immunoglobulin to 40 CHF patients during a small double blind, placebo controlled trial increased plasma concentrations of IL-10, which eventually yielded improvements in left ventricular ejection fraction (LVEF) (from 26 (2)% at baseline to 31 (3)% after treatment, p < 0.01). 15 IL-10 and other substances augmenting its production may offer therapeutic possibilities in CHF.
C REACTIVE PROTEIN
C reactive protein (CRP) was first discovered in 1930. It was so named because it reacts with the somatic C polysaccharide of Streptococcus pneumoniae. CRP specifically binds to specific microbial polysaccharides (phosphocholine moieties), which gives this substance a host defensive role. Upon binding to these structures, CRP activates the classical complement pathway and opsonises ligands for phagocytosis. CRP is exclusively produced in the liver. It is secreted in increased amounts within six hours of an inflammatory stimulus and is therefore regarded as a marker of acute inflammation.
The first observation of raised concentrations of CRP in CHF was published in 1990. Recently, another group measured CRP values in 188 patients with idiopathic dilated cardiomyopathy.16 All patients had an impaired LVEF of less than 40%. Those patients who died during a follow up period of five years had significantly higher CRP concentrations than those who survived (1.05 (1.37) mg/dl v 0.49 (1.04) mg/dl, p < 0.05). Moreover, CRP is able to augment IL-1β induced production of iNOS, although NF-κB production is not altered. However, it is not clear if CRP is merely a marker of inflammation with no particular role in the development of cardiac disease or if it directly modulates disease process.
ADHESION MOLECULES
Adhesion molecules are cell surface receptors involved in the binding of leucocytes to each other, to endothelial cells, or to the extracellular matrix (fig 3⇓). These molecules have been implicated in a vast number of cardiovascular diseases. Three different families of proteins have been described so far: (1) The immunoglobulin superfamily consists of a number of adhesion molecules including intracellular adhesion molecule-1 (ICAM-1), ICAM-2, ICAM-3, vascular cell adhesion molecule-1 (VCAM-1), and others. (2) Integrins form the counterreceptor to the latter type of receptor, mediating leucocyte adherence to the vascular endothelium and other cell–cell interactions. Integrins are heterodimers of α and β subunits. Among them, lymphocyte function associated antigen-1 (LFA-1) and glycoprotein IIb/IIIa can be found (fig 3⇓). (3) The selectins are involved in the adhesion of leucocytes to activated endothelium. These weak interactions cause a typical “rolling” of leucocytes on the endothelial surface, which is mainly mediated by leucocyte (L)-selectin and platelet (P)-selectin (fig 3⇓). Other selectins, such as endothelial (E)-selectin, seem to produce a stronger interaction, which eventually leads to cell extravasation. Since immune activation is an important finding in CHF, it is tempting to speculate that the interaction between endothelial cells, leucocytes, and possibly platelets is of particular importance in this condition.

{kind=link}
{kind=link}
{kind=link}
Physiological role of different types of adhesion molecules. Selectins are mainly involved in leucocyte rolling on the endothelial wall. Integrins—for example, LFA-1—yield a much stronger interaction, which eventually leads to transendothelial migration. Endothelium derived NO plays another important role in mediating endothelial function. Its production by NOS from L-arginine leads to smooth muscle relaxation. cNOS, constitutive nitric oxide synthase; ICAM, intracellular adhesion molecule; LFA, lymphocyte function associated antigen; NO, nitric oxide; PECAM, platelet endothelial cell adhesion molecule.
An upregulation of plasma soluble ICAM-1 has been shown in a group of 102 CHF patients.17 The values in this study increased from the normal concentration (149 (10) ng/ml) to 207 (9.4) ng/ml in mild CHF, and 293.18 ng/ml in severe CHF (all p < 0.05). Moreover, a significant negative correlation between LVEF and soluble ICAM-1 was observed (r = −0.36; p < 0.001), and these values independently predicted survival. Elevated serum concentrations have also been demonstrated for other soluble markers, such as VCAM-1, P-selectin, and E-selectin.4,18 Interestingly, only VCAM-1 concentrations decreased after heart transplantation.
The failing myocardium itself displays an increased expression of ICAM-1 and LFA-1.19 TNFα induces adhesion molecules. Thus, this cytokine may account for mononuclear cell infiltration into the myocardium, which has been reported in CHF.
NITRIC OXIDE
Nitric oxide (NO) was originally discovered in 1980 by Furchgott and Zawadski. It is a lipophilic, freely diffusible, soluble gas, which has a short half life of less than four seconds in biological solutions. Its nearly ubiquitous involvement has resulted in an explosion in the NO field in the last years. NO is produced from the amino acid L-arginine by nitric oxide synthase (NOS) (fig 3⇑), and it reacts with O2 in aqueous solutions yielding the relatively inert nitrate (NO3−) and nitrite (NO2−). However, NO also reacts with oxygen derived free radicals, namely superoxide anion, to form the toxic peroxynitrite (ONOO−). NO is produced by a group of well characterised isoforms of nitric oxide synthase (NOS). Three isoforms have been identified. (1) The endothelial (constitutive) isoform (cNOS, also known as NOS3) produces a continuous amount of NO, which acts as a vasodilator. In endothelial cells, cNOS is mainly found in the membrane of caveolae, small invaginations, which are characterised by the presence of a marker protein termed caveolin. After synthesis in the endothelium, NO diffuses across the cell membrane and enters vascular smooth muscle cells to induce muscle relaxation (fig 3⇑). (2) The inducible isoform of NOS (iNOS or NOS2) has already been mentioned. Unlike membrane bound cNOS, iNOS is found in the cytoplasm of a large number of different cell types. After induction of iNOS by, for example, pro-inflammatory cytokines, interferon γ or LPS, the output of iNOS far exceeds that of cNOS. Since the intracellular NO content of macrophages rises during bacterial infection, NO is thought to play an important role in microbial killing in innate immune responses. (3) The third isoform of NOS is the neuronal isoform nNOS (NOS1). It is expressed in the central and peripheral nervous systems and in skeletal muscles.
The role of NO in CHF is complex. On the one hand, lack of NO is leading to endothelial dysfunction with its detrimental consequences including impaired tissue perfusion, myocardial ischaemia, and vascular remodelling. On the other hand, higher concentrations of NO, which have been observed in the failing myocardium, may cause the loss of myocytes and inhibit myocyte contractility.20 While low (physiological) concentrations of NO have beneficial effects, those of higher (pathological) concentrations, possibly mediated by iNOS, appear detrimental. NO induced by cytokines also has negative chronotropic effects and is capable of triggering apoptosis. Indeed, high iNOS gene expression is associated with low LVEF, but not with clinical disease severity of heart failure.21 Another study described a linear relation between iNOS gene expression and LVEF.22 In the light of this finding it is not surprising that L-NG-monomethyl-arginine (L-NMMA), an NOS inhibitor, blocks negative inotropic effects. The different mechanisms by which NO results in these contrasting effects seen in CHF may involve decreases and increases in oxidative stress, respectively.
LEUCOCYTE SUBSETS IN CHF
Little has been published about leucocytes and their functions in the course of CHF. This is fairly surprising as it is commonly believed that CHF is a state of chronic immune activation. In one interesting analysis, myocardial tissue was obtained from patients with end stage heart failure at transplantation.19 Interestingly, CD3+ T lymphocytes and CD68+ macrophages were more abundantly present in the myocardium of CHF patients than in controls, suggesting a role of adhesion molecules in this setting. Indeed, this may significantly contribute to the structural deterioration that is the basis of reduced cardiac function in this setting. A low lymphocyte count, however, seems to be rather detrimental. On the other hand, the B cell response seems to be intact with normal antibody secretion capacity upon pneumococcal vaccination.23 Another study reported a decreased percentage of T suppressor cells in patients with idiopathic dilated cardiomyopathy. The significance of this finding remains to be elucidated.
THERAPEUTIC OPTIONS FOR CHF
The prognosis of CHF remains fairly poor, although the introduction of angiotensin converting enzyme (ACE) inhibitors and β blockers improved survival significantly. Recent insights into the pathophysiology of CHF provide promising targets for future treatments. However, preliminary results from the first large scale clinical trial to counterbalance TNFα activity have been rather disappointing. Insights into other possible therapeutic targets are only about to begin.
TNFα antagonism with etanercept and infliximab
Only recently, two trial programmes to investigate the therapeutic impact of TNFα antagonism in vivo have been halted. These trials tested two different approaches to attenuate the detrimental effects of TNFα.
Etanercept is a TNFR-2 fusion protein, which binds to TNFα and functionally inactivates this cytokine. A small double blind, placebo controlled pilot study in 18 patients with moderate heart failure showed promising results.24 Thus, a large scale clinical trial was designed with two arms. Both the American arm RENAISSANCE (randomized etanercept North American strategy to study antagonism of cytokines) and the European arm RECOVER (research into etanercept: cytokine antagonism in ventricular dysfunction) recruited a total of 2048 CHF patients, most of them with mild to moderate disease. The combined analysis of the two trials was termed RENEWAL (randomized etanercept worldwide evaluation). Patients were treated subcutaneously for 24 weeks with etanercept or placebo in a double blind fashion (doses in RECOVER: 25 mg two times weekly or 25 mg once weekly; doses in RENAISSANCE: 25 mg three times weekly or 25 mg two times weekly). For reasons that are not entirely clear, the RENEWAL analysis excluded the lowest dose of RECOVER (that is, etanercept 25 mg once weekly). The number of patients classified “improved”, “unchanged”, or “worsened” was similar for patients on placebo or any dose of etanercept. Moreover, the primary end point (death or hospitalisation because of CHF) was not different between the two groups (risk ratio (RR) 1.10, 95% confidence interval (CI) 0.91 to 1.33; p = 0.33).25 The secondary end point (all cause mortality) did also not differ between the two groups (RR 1.13, 95% CI 0.86 to 1.50; p = 0.39). The survival curves overlapped throughout the first year of treatment. Since the combined analysis of the two trial programmes excluded the low dose group from RECOVER, it appears noteworthy that the adjusted effects on total mortality for the individual dose levels were not reported. Of particular interest is total mortality in the 375 patients on 25 mg etanercept once weekly compared with the 373 patients on placebo in RECOVER. The unadjusted mortality rates in RECOVER were 8.8% in the placebo group, 5.9% for etanercept once weekly and 7.2% for etanercept twice weekly. In other words, the total mortality of patients treated with low dose etanercept was more than 30% reduced compared to the placebo group. In light of these data, a so far neglected dose dependent effect may substantially change the interpretation of the studies.26
Inflammatory mediators in CHF: key points
Immune activation plays an important role in the disease progression of chronic heart failure (CHF)
Pro-inflammatory cytokines, especially that of TNFα, IL-6, and IL-1, exacerbate haemodynamic abnormalities, exert directs toxic effects on the heart, and contribute to tissue wasting and weight loss in CHF (that is, cachexia). The origin of pro-inflammatory cytokines in CHF is not entirely clear
Endotoxin (LPS) may play an important role in the process of activating pro-inflammatory cytokines
Adhesion molecules may contribute to leucocyte extravasation in the myocardium, thus promoting structural deterioration as a basis of reduced cardiac function
Recent clinical trials to inhibit TNFα activity in CHF showed disappointing results. It seems likely that anti-cytokine therapy will work only in CHF patients with proven inflammatory status. Statins, because of their pleiotropic (anti-inflammatory) effects, may prove beneficial in this setting
The ATTACH trial (anti-TNFα therapy against chronic heart failure) enrolled 150 patients with CHF to investigate the impact of treatment with infliximab, a chimeric (mouse/human) IgG1 monoclonal antibody that binds both soluble and membrane bound TNFα.27 It is administered intravenously. Like RENAISSANCE and RECOVER, this study was designed in a multicentre, randomised, double blind, placebo controlled fashion.27 It has also been stopped prematurely. However, when analysing the data from ATTACH it is noteworthy that the plasma values of infliximab achieved in the patients were many times higher than expected. This may account for the increased risk of death in the group receiving the higher dose of infliximab (10 mg/kg body weight) observed in this study (RR 2.84, 95% CI 1.01 to 7.97; p < 0.05). There was no adverse risk associated with 5 mg/kg body weight infliximab (RR 0.80, 95% CI 0.22 to 2.99) and in fact in patients receiving this dose LVEF improved (p < 0.05).27
The lesson learned from these trials is that TNFα may not only exert detrimental effects. However, some questions remain to be addressed. Both studies included only a very limited number of patients with severe CHF who can be expected to have significant inflammatory immune activation. Interestingly, ATTACH aimed to recruit patients with severe CHF, but none of the 49 patients in the placebo group died during the first 28 weeks of follow up.25
Statins
Statins (3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors) block the rate limiting step in cholesterol biosynthesis in the liver and other tissues. These drugs are administered orally, are well tolerated, and generally safe. It has been reported that statins can improve the prognosis of coronary artery disease irrespective of serum cholesterol values, which gave rise to the idea that effects beyond cholesterol lowering—so called pleiotropic effects—exist.
Indeed, some substances from this group have been shown to improve endothelial function by inducing cNOS gene transcription.28 Some statins might be able to reduce vascular production of reactive oxygen species. Moreover, statins have been found to reduce C reactive protein values after myocardial infarction and in hypercholesterolaemia. Moreover, statins might decrease the production of TNFα, IL-1, and IL-6 from macrophages.29
The availability of clinical data of statin use in CHF is scarce. The Scandinavian simvastatin survival study (4S) demonstrated fewer instances of new onset CHF after simvastatin treatment. This effect seems to be mediated by the pleiotropic effects of the drug, because changes in the lipoprotein profiles and baseline characteristics were similar in patients with or without future events. The clinical benefit associated with statin treatment was also independent of baseline cholesterol values in WOSCOPS (West of Scotland coronary prevention study). A subgroup analysis revealed that cholesterol independent mechanisms may provide additional benefits. Prospective data are needed to examine the effects of statin treatment in CHF. The doses needed to achieve the desired effects might be much lower than those needed to treat hypercholesterolaemia.29
CONCLUSION
The last several years have seen considerable research into the field of immune activation in CHF. Pro-inflammatory cytokines have been implicated in the progression of heart failure. Several hypotheses were suggested to explain the development of this aspect of CHF. The role of leucocytes is not entirely understood; however, it appears that raised concentrations of adhesion molecules indicate a role for these molecules in the extravasation into the myocardium and other tissues. The role of LPS in triggering inflammation needs further study. The disappointing results of recent trials most likely will lead to slow down of drug development processes. We believe the “cytokine story” is still very much alive in CHF. However, anti-inflammatory strategies need to be broad and focused on patients with inflammatory problems. New therapeutic developments will hopefully lead to improvements in the still poor prognosis of CHF patients.
Acknowledgments
SDA is supported by a Vandervell Fellowship and a donation from Dr Hubert Bailey. Applied Cachexia Research (SDA) is supported by a grant of the Charité Medical School. SvH is supported by the German Heart Foundation.
REFERENCES
Footnotes
↵* Also Applied Cachexia Research, Department of Cardiology, Charité Medical School, Campus Virchow-Klinikum, Berlin, Germany
Linked Articles
- Miscellanea
- Miscellanea